Parts Group
Understanding human DNA function by engineering
Research overview
Approach
The following four statements describe our approach:
1) We get things done. We start projects with clearly defined goals, and publish both positive and negative results of the ones that pass the pilot stage. We deliver to our collaborators.
2) We work on important problems. We pick projects based on how much they impact our understanding of human cells, characterize the variation of gene function across individuals, or influence how others work.
3) We succeed as a team. We have a diverse mix of backgrounds and skillsets, complementing each other with our strenghts.
4) We are excited about science. We read broadly, discuss latest developments, and keep up to date both with the depth of our field, and the entire breadth of genomics.
Projects
Deciphering genomes through engineered structural variation
 that affect sequence at the scale needed to interrogate gigabases of the human genome.
that affect sequence at the scale needed to interrogate gigabases of the human genome.Predicting gene editing outcomes

Large scale screens of coding variant effects in humans
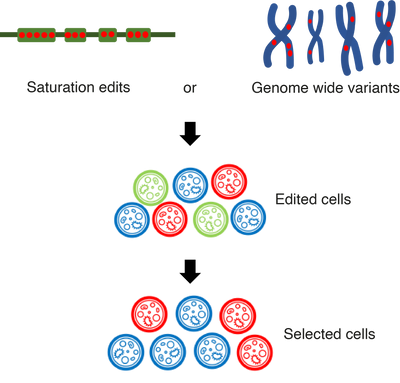 We are interested in using genome editing tools, such as base and prime editing, for large scale pooled variant effect screens in human cells and comparing their properties to similar screens using established techniques such as saturation genome editing (SGE). These screens involve generating a pool of cells carrying a library of variants and then subjecting them to a selection pressure related to the gene of interest, which can be intrinsic or artificial, for instance linking activity to fluorescence.
We are interested in using genome editing tools, such as base and prime editing, for large scale pooled variant effect screens in human cells and comparing their properties to similar screens using established techniques such as saturation genome editing (SGE). These screens involve generating a pool of cells carrying a library of variants and then subjecting them to a selection pressure related to the gene of interest, which can be intrinsic or artificial, for instance linking activity to fluorescence. Finding sequence determinants of context-specific regulatory elements
 measure the regulatory activities of sequences at scale. Using this data, we can more systematically analyse how smaller, more controlled DNA changes lead to different regulatory activities. We are exploring how we can engineer new regulatory elements with specific context-specific activities using machine learning models. We hope that this toolbox expands our understanding of how genes are regulated by their sequence context.
measure the regulatory activities of sequences at scale. Using this data, we can more systematically analyse how smaller, more controlled DNA changes lead to different regulatory activities. We are exploring how we can engineer new regulatory elements with specific context-specific activities using machine learning models. We hope that this toolbox expands our understanding of how genes are regulated by their sequence context.Modifiability of CRISPR perturbation effects in iPSCs with scRNA-seq and growth assays
 characterize cell-specific gene function, identify protein complex membership and predict causal genes. Unlike previous studies, which generally establish proof-of-concept by demonstrating knockout in a handful of genes or focus on a handful of cell-lines, we are performing these experiments at scale by knocking down thousands of genes across hundreds of individuals to then identify role of genetic background in phenotype penetrance.
characterize cell-specific gene function, identify protein complex membership and predict causal genes. Unlike previous studies, which generally establish proof-of-concept by demonstrating knockout in a handful of genes or focus on a handful of cell-lines, we are performing these experiments at scale by knocking down thousands of genes across hundreds of individuals to then identify role of genetic background in phenotype penetrance.
Core team

Dr Alistair Dunham
Postdoctoral Fellow

Mr Gareth Girling
Advanced Research Assistant

Elin Madli Peets
Advanced Research Assistant

Isabelle Zane
PhD Student
Previous core team members

Dr Felicity Allen
Postdoctoral Fellow

Luca Crepaldi
Staff Scientist

Jacob Hepkema
PhD Student

Dr Michelle McRae
Senior Research Assistant/Laboratory Manager

Danesh Moradigaravand
Senior Bioinformatician

Dr Daniele Muraro
Senior Bioinformatician

Kasia Tilgner
Visiting Scientist

Juliane Weller
PhD Student

Dr Yan Zhou
Postdoctoral Fellow
Related groups
Programmes and Facilities
Partners
We work with the following groups
External
Cancer Dependency Map
The Cancer Dependency Map aims to find a targetable dependency in each cancer cell.