cell2location
Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics (cell2location model)
About

cell2location leverages reference cell type signatures that are estimated from scRNA-seq profiles, for example as obtained using conventional clustering to identify cell types and subpopulations followed by estimation of average cluster gene expression profiles. Cell2location implements this estimation step based on Negative Binomial regression, which allows to robustly combine data across technologies and batches. Using these reference signatures, cell2location decomposes mRNA counts in spatial transcriptomic data, thereby estimating the relative and absolute abundance of each cell type at each spatial location (see figure below).
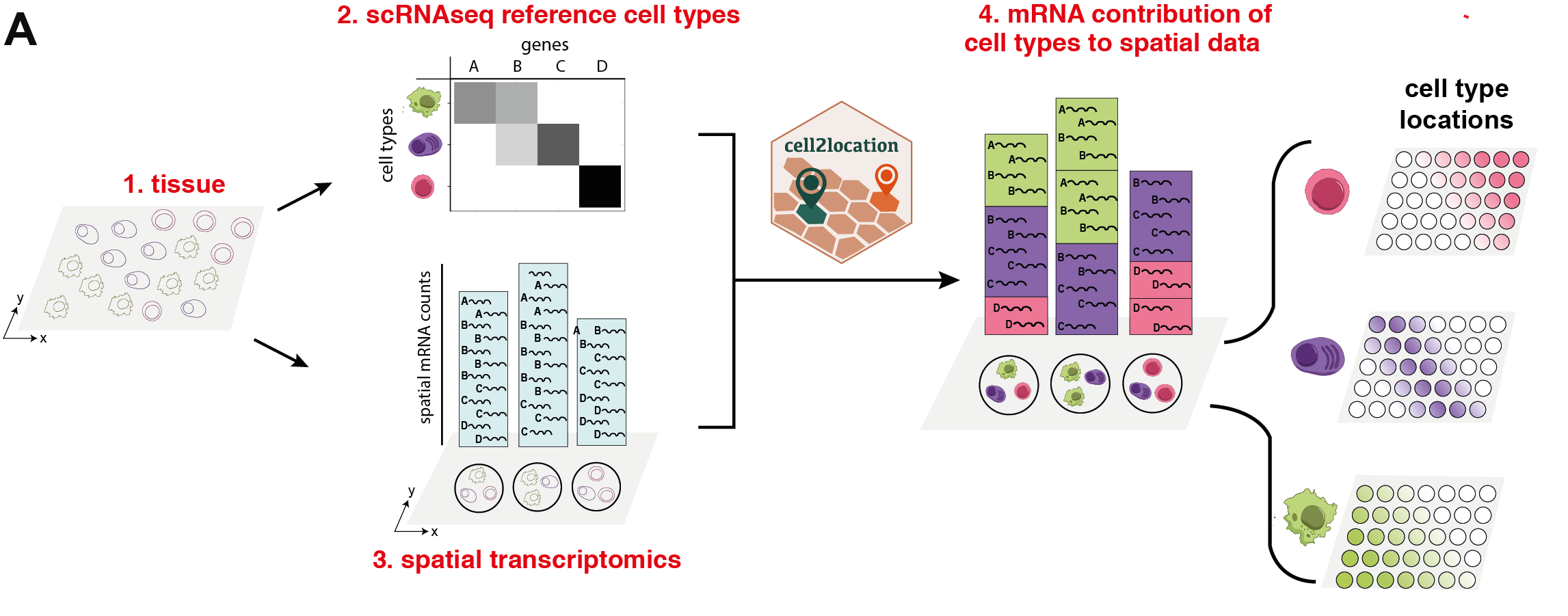
Cell2location is implemented as an interpretable hierarchical Bayesian model:
- providing principled means to account for model uncertainty;
- accounting for linear dependencies in cell type abundances;
- modelling differences in measurement sensitivity across technologies;
- accounting for unexplained/residual variation by employing a flexible count-based error model.
Cell2location is computationally efficient, owing to variational approximate inference and GPU acceleration. For full details and a comparison to existing approaches see our preprint https://www.biorxiv.org/content/10.1101/2020.11.15.378125v1.
The cell2location software comes with a suite of downstream analysis tools, including the identification of groups of cell types with similar spatial locations.
Downloads
There are 2 ways to install and use our package: setup your own conda environment or use the singularity and docker images (recommended). See below for details.
You can also try cell2location on Google Colab on a smaller data subset containing somatosensory cortex.
Please report bugs via https://github.com/BayraktarLab/cell2location/issues and ask any usage questions in https://github.com/BayraktarLab/cell2location/discussions.
We also provide an experimental numpyro translation of the model which has improved memory efficiency (allowing analysis of multiple Visium samples on Google Colab) and minor improvements in speed – https://github.com/vitkl/cell2location_numpyro. You can try it on Google Colab. However, note that both numpyro itself and cell2location_numpyro are in very active development.
Further information
Usage and Tutorials
Tutorials covering the estimation of expression signatures of reference cell types (1/3), spatial mapping with cell2location (2/3) and the downstream analysis (3/3) can be found here: https://cell2location.readthedocs.io/en/latest/
The architecture of the package is briefly described here. Cell2location architecture is designed to simplify extended versions of the model that account for additional technical and biologial information. We plan to provide a tutorial showing how to add new model classes but please get in touch if you would like to contribute or build on top our package.
Sanger Institute Contributors

Dr Anna Arutyunyan
Postdoctoral Fellow

Dr Omer Bayraktar
Group Leader

Dr Tong LI
Senior Software Developer


Dr Lauma Ramona
Research Administrator Tree of Life Programme

Dr Oliver Stegle
Associate Faculty in the Cellular Genomics Programme

Roser Vento-Tormo
Group leader
Previous contributors

Dr Vitalii Kleshchevnikov
Bioinformatician

Dr Jun Sung Park
Visiting Scientist
External Contributors

Artem Shmatko
EMBL-EBI and Moscow State University
Artem Lomakin
EMBL-EBI
Mika Sarkin Jain
University of Cambridge
Liz Tuck
Sanger Institute
Moritz Gerstung
EMBL-EBI
Louisa James
Queen Mary University of London
Luz Garcia Alonso
Sanger Institute