Jaron Group
Evolution of unusual reproductive mechanisms
Our work
One of the oldest riddles of evolutionary biology is why there is such an amazing diversity of reproductive strategies. We are interested in quantifying this diversity, understanding where it comes from and what roles do the method of reproduction (sexual, asexual, other forms), structural changes in the chromosomes, and genetic variation in the DNA sequence itself play in driving or tempering evolution.
To achieve this we will apply genomic analysis at a vast scale, across many species to identify the genomic events that are strongly associated with the move to, and ongoing success of, specific reproductive strategies.
Because the majority of studies that have been carried out so far have focused on individual species or an individual branch of the evolutionary tree, we have only been able to identify the genomic changes that have been acquired over that species’ evolutionary history. While this work has produced useful insights, it cannot reveal which genomic events are strongly associated with a particular reproductive approach, and which events are coincidental.
By looking at the bigger picture, our team will be able sift out coincidental genomic events. We will read and analyse the genomes of many different species and evolutionary branches to survey the evolutionary forces at work across species at scale. This approach will enable us to discover those events and genomic changes that are shared across species with the same reproductive strategies, and separate out those coincidental events that occur infrequently.
Our moonshot is to study all species that share the same reproductive strategy at the same time!
We look to disentangle the individual forces at work by studying the genomes of species with various forms of non-Mendelian inheritance: for example, those whose reproduction is asexual, or genomes with parental bias in transmission (e.g. Y chromosomes). We hope that comparing the genomes and genomic events at play in these species with those undergoing Mendelian inheritance will reveal genetic elements and chromosome rearrangements that drive evolution and adaptation: the universal genetic and genomic ‘rules’ of evolution.
To achieve this, our team is working closely with our colleagues in the Darwin Tree of Life project to systematically sequence and characterise all the peculiar chromosomes the project uncovers. We are creating standardised, scalable processes, pipelines and analysis methodologies to survey within-species karyotype diversity of numerous species of interest and extract chromosomal assignments from the sequencing data at scale.
Our aim is to create the foundational tools, processes and insights that will go beyond being published papers, and will become the bedrock resources that enable researchers around the world to use genomics to study evolution of reproduction.
Sex chromosome evolution
Much of genomic research has focused on the roles of selection, linkage and dominance in influencing evolution. However, these forces are closely intertwined, and it is very hard to untangle the individual effect that each plays in driving adaptation and speciation.
By studying different forms of reproduction, where some (but not all) aspects of inheritance typical for sex chromosomes, chromosome recombination and chromosome segregation are altered, we can start to discern influence each element effects.
One example of this is X chromosome evolution. Its inheritance (and so its evolution over generations) is different to the other chromosomes in the genome. Only one copy is (usually) passed on to males, while two copies are passed on to females. So, males will display any recessive traits carried on their inherited X chromosome, while females can hide them by the second chromosome copy. In addition, in comparison to autosomes, X chromosomes spend much more time in females (up to 2/3rds compared with 1/2 in males). And males will pass the X chromosomes to female offspring only, while females will pass theirs to offspring of both sexes.

By studying X chromosome inheritance and evolution across many members of the same species and across taxa, we will seek to understand these forces at the genomic level.
In particular, we are focussing on the evolution of globular springtails. These creatures not only have large X chromosomes, they also have paternal gene elimination so that all males’ sperm only carry the female haplotype. This means that the X chromosomes are passed from generation to generation in exactly the same way as all other non-sex chromosomes, removing the usual confounding factors at work. This allows us to compare X chromosome inheritance and genomic alterations with ‘standard’ chromosomes unhindered by outside influences.
Mapping the full genome diversity of UK-based species and how this affects genomic evolution
Across the 70,000+ species being sequenced by the Darwin Tree of Life project the diversity of genomes is fascinating and staggering. The genomes have different ploidies, are small or large, stable, or dynamic, completely homozygous or extremely heterozygous. By harnessing these differences, we seek to understand the causal effects between evolutionary forces (such as effective population sizes, recombination rates, and sexual or intragenomic conflicts) and genetic changes.
To achieve our goal, our team compares genomes and chromosomes from species with various types of non-Mendelian inheritance such as:
- the sex chromosomes of species
- B chromosomes
- germ-line restricted chromosomes of species
- haplodiploids
- recombination and segregation in parthenogenesis (species that can develop embryos from their eggs without fertilisation by sperm).
Previously such comparisons were carried out on a single transition and were subject to confounding idiosyncrasies specific to that species and moment of transition. To overcome this, our team conducts large-scale comparisons of the genomic features found in species captured by the Darwin Tree of Life project with reference to their form of reproduction.
A key foundational part of this work is building and deploying a systematic, scalable approach using k-mer comparison to annotate all unusual chromosomes of interest and link this genetic information with vital species-specific metadata regarding the reproductive mode and/or non-canonical chromosomal constellation.
Creating a database of reproductive metadata and genome constitution for the research community
We are validating the process by utilising the chromosome size and constitution data gathered from previous flow cytometry measurements and existing cytology papers.
We are keen to work with reproductive biologists around the world to create a community-driven curation system that brings together existing reproductive metadata from published literature with existing Darwin Tree of Life resources such as Genomes on a Tree. This resource will provide a vital foundation for global research endeavours.
If you are interested in collaborating with us on building this foundational resource for the researchers worldwide, please email Kamil Jaron.
Capturing genome complexity using sequencing data
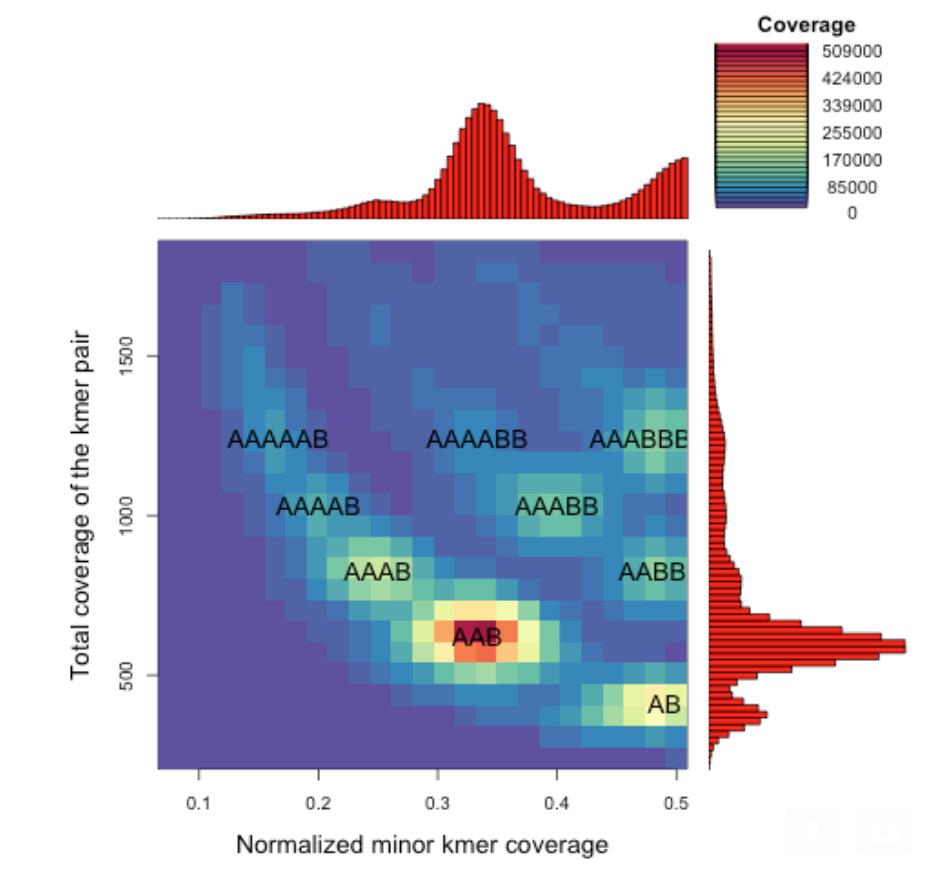
In addition, we will be looking to develop a range of tools to discover the genomic structures and features associated with reproductive strategies.
For example, one confounding factor in evolution research using genomics is the variations in the number of copies of the chromosomes in the nucleus (ploidy). Using both techniques described above and enhancements to our smudgeplot software, we seek to identify polyploid genomes across the Darwin Tree of Life.
Join our team
We are always on the lookout for bright and motivated people to join the group so, if you’re interested, please send Kamil an email and include your CV.

Core team

Dr Aleksandra Bliznina
Postdoctoral Fellow

Sam Ebdon
Postdoctoral Fellow

James McCulloch
PhD Student
Related groups
Partners
We work with the following groups