Genome Engine
Tree of Life aims for the highest standard of genome assemblies wherever possible. This means complete genome sequences that span each chromosome of the nuclear genome (the DNA in the nucleus), plus the genetic material in all organelles (e.g. mitochondria and chloroplasts). Genes are then identified and features annotated to help users make sense of how different parts of the sequence function.
Our aim
The Tree of Life programme wants the next generation of scientists to operate in a genome-ready world. Currently, a typical PhD researcher might spend up to a year sequencing the genome for their chosen species. This is a quarter to a third of their project which would be better spent probing the questions they set out to study – if only the relevant reference genome were available.
Once a reference genome is available, a whole toolkit of other exciting methods and techniques are unlocked. For example, scientists can use resequencing to look at the DNA of other individuals within the same species and compare back to the reference genome. In this way, it becomes quicker, cheaper and more efficient to study organisms’ biology and evolution, support conservation efforts, or search for new biomedicines and other compounds.
Producing a chromosomally complete genome
We have built an end-to-end, sample-to-genome pipeline – the Genome Engine – for producing chromosomally-complete genome sequences.
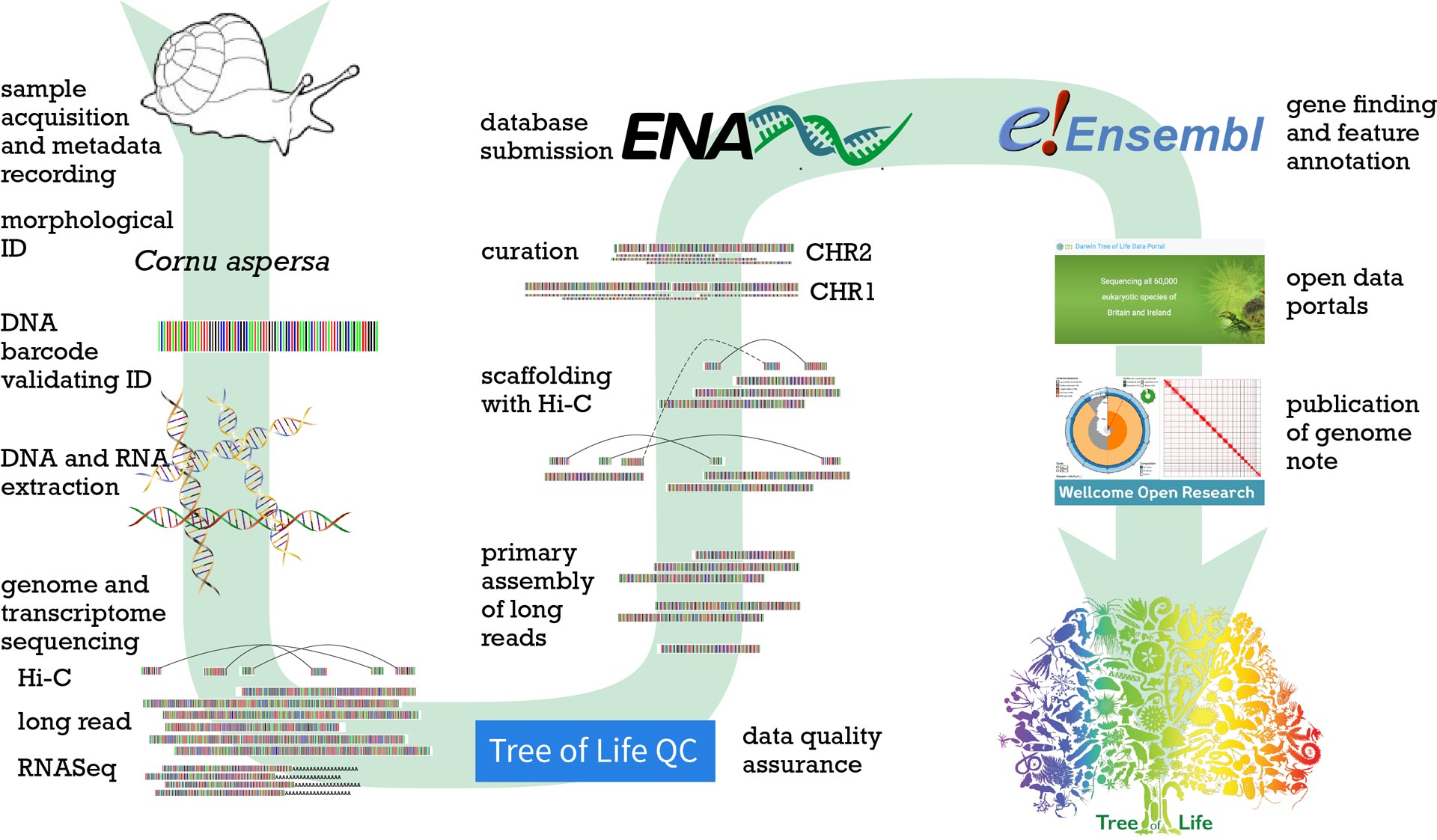
Sample acquisition and metadata collection
Our collaborators first collect and identify the species. This follows detailed standard operating procedures that ensure that collections are both legal and ethical. We work closely with the Sanger’s Legal and Governance teams to ensure that all permissions and in particular Nagoya Protocols permits and CITES documentation for samples coming from overseas and/or from endangered species – are in order before shipping. We coordinate our work with other projects – avoiding overlaps and building synergies – in the Earth BioGenome Project through a data system developed in ToL called Genomes on a Tree (goat.genomehubs.org).
Morphological identification
We rely on our expert colleagues in our biodiversity partner institutions,a nd in the wider expert amateur and professional taxonomy community, to identify the specimens. This is not always an easy task: there are many, many species of small flies which, to an untrained observer, all look the same.
DNA barcoding
In DNA barcoding, we sequence a standardised, small piece of the genome of each specimen and use this sequence both to confirm the identification and to track the samples as they work their way through our systems. With over 10,000 species and 200,000 samples, tracking where a species is in its genome journey and where each tube is is very important. Again we have built a dedicated Sample Tracking System and the ToL Portal (https://portal.tol.sanger.ac.uk/) to assure ourselves that all is proceeding to plan.
DNA and RNA extraction
In the Tree of Life Core Laboratory and the Long Read Extraction Service in Scientific Operations, we use carefully selected methods to extract ultra high molecular weight (i.e. very long) DNA from each sample. Extraction methods are tailored to the special characteristics of each species – whether stony corals, minute nematodes, or tough plants. We make our methods available openly (see protocols.io ToL workspace at https://www.protocols.io/workspaces/wellcome-sanger-institute13).
Genome and transcriptome sequencing
The DNA material is then sequenced using the latest long-read technologies – we use both the PacBio Revio and the Oxford Nanopore Technologies Promethion platforms. We also generate special long-range information about which fragments are likely to be on the same chromosome – called Hi-C sequencing.
We also generate sequence data from the RNA of each species. This information tells us which parts of the genome are expressed as messenger RNA and thus translated into proteins.
Data quality assurance
It is essential that the data we generate is of the highest quality, and thus we interrogate the raw genome data with a series of tools, many written by ToL and Sanger informaticians. These data are displayed on an open-access web portal, ToLQC (https://tolqc.cog.sanger.ac.uk/), so that collaborators outside the Sanger can keep track of progress in “their” organism.
Primary assembly
The long read data is first “assembled” into a primary set of contiguous sequences, or contigs by the Tree of Life Assembly (ToLA) team. This process involves the use of dedicated high performance computers and a set of innovative computer programmes, some written by ToL and Sanger informaticians. We also assemble the genomes of the organelles of each species – the chloroplasts of plants, and the mitochondria found in (nearly) every species.
Scaffolding
ToLA then use the Hi-C data to “scaffold” the primary assembly contigs into longer sequences, which may still contain some “gaps” where our tools are unable to predict what the true sequence should be.
Curation
All the genomes ToL releases have been curated by expert scientists in our Genome Reference Informatics (GRIT) team, who check that each sequence is in the right place and in the correct orientation. The GRIT team also checks that we have not inadvertently included sequence form any of the microbes or other contaminants that come with the wild-caught samples.
These ToL informatics teams are hugely skilled at building repeatable pipelines of these bioinformatics workflows, and publish/release them openly (see https://pipelines.tol.sanger.ac.uk/).
Database submission
All ToL data are submitted to the public archives rapidly (on completion of assembly) and openly. We deposit data in the European Nucleotide Archive, from where it is shared round the world by the International Nucleotide Sequence Database Collaboration (INSDC).
Gene finding and feature annotation
Colleagues in the European Bioinformatics Institute Ensembl team generate annotations of the genomes. They use the transcriptome data to identify where the protein-coding genes are in the sequence, and identify repeat sequences and other features. These annotations are published immediately and openly on Ensembl.
Open data portals
Our colleagues at the European Bioinformatics Institute then aggregate all the information about the genomes (the sample metadata collected when the specimen was collected and all the raw and assembled data) and present them through portals such as portal.darwintreeoflife.org and portal.aquaticsymbiosisgenomics.org.
Publication of genome note
Finally we write a short publication, written by the collectors, and taxonomists as well as the genome scientists at Sanger and EBI, that introduces the species, describes the process used to sequence it, summarises the data and the assembly quality, and announces that the genome is open for all to use. These Genome Notes are published at Wellcome Open Research (https://wellcomeopenresearch.org/gateways/treeoflife).