Behjati Group
Connecting human development and disease
Our team uses cutting edge single cell techniques to understand normal human development and how normal development relates to disease. We focus on understanding and quantifying the nature and origins of cancer cells, particularly childhood cancers. To do this we combine the study of somatic genomic changes, bulk and single cell transcriptomics, and mathematical modelling of data.
We understand where most cancers arise at a broad anatomical level (e.g. nephroblastoma develops in the kidney), but not at a precise cellular level. Furthermore, we do not have a clear understanding of what normal transcriptional profiles cancer cells are most reliant on. Our research aims to improve our understanding of cancer cell’s transcriptomic nature and precise cell of origin. This broad aim is split across three complementary study areas:
- Reconstruction of cancer cell of origin using somatic mutations
- Matching transcriptomic profiles of diseased cells with normal tissues transcriptomes
- Distilling clinically relevant features of disease transcriptomes
Cancer cell of origin
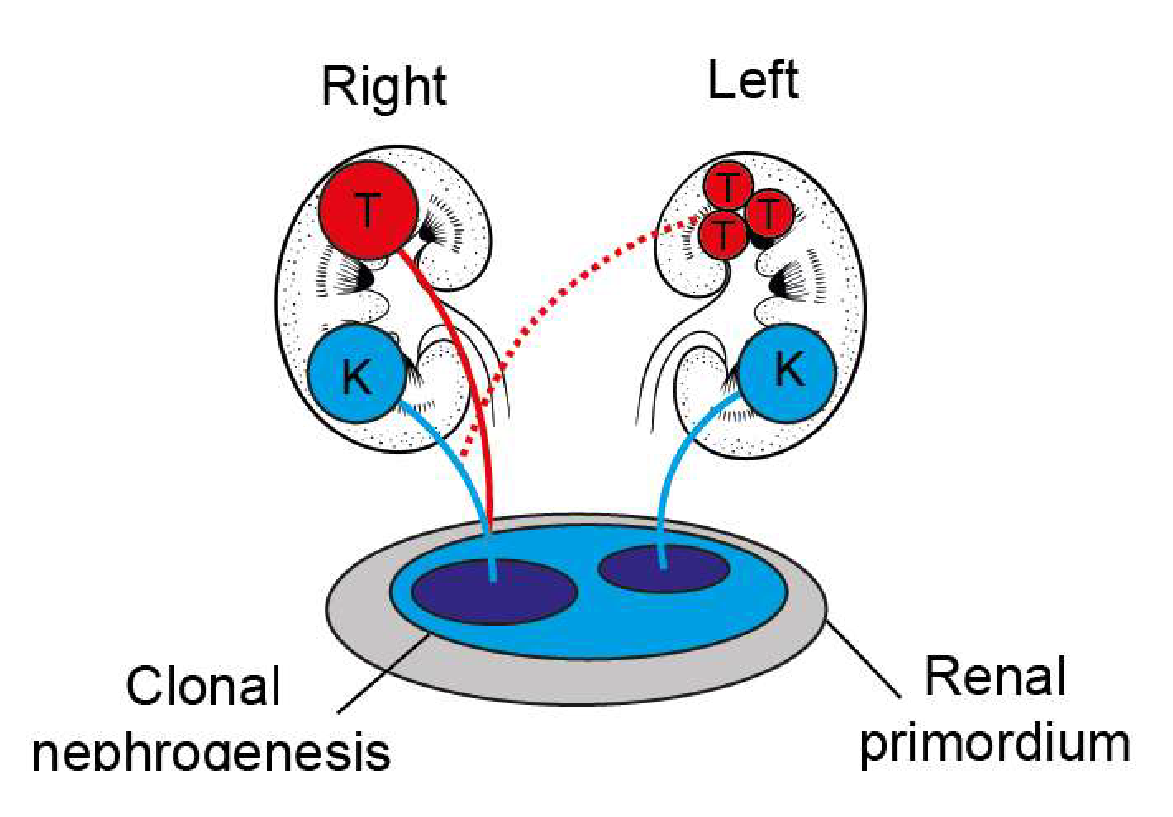 Definitive proof of a cancer’s cell of origin can only be provided by tracing the cancers lineage. That is, to know which cell a tumour arises from, we must construct a phylogenetic tree relating normal tissues to cancer cells. Each cell division results in the accumulation of ~2 somatic mutations across the entire genome. These random somatic mutations can be used as markers and permit the relationship between different cells and tissues to be reconstructed. Specifically, by taking multiple biopsies from the same individual, we can use whole genome sequencing to identify which somatic mutations are shared between cancer cells and different normal tissues. An example of the application of this approach is our work identifying that Wilms tumour develops from kidneys with an unusually high level of clonality (clonal nephrogenesis).
Definitive proof of a cancer’s cell of origin can only be provided by tracing the cancers lineage. That is, to know which cell a tumour arises from, we must construct a phylogenetic tree relating normal tissues to cancer cells. Each cell division results in the accumulation of ~2 somatic mutations across the entire genome. These random somatic mutations can be used as markers and permit the relationship between different cells and tissues to be reconstructed. Specifically, by taking multiple biopsies from the same individual, we can use whole genome sequencing to identify which somatic mutations are shared between cancer cells and different normal tissues. An example of the application of this approach is our work identifying that Wilms tumour develops from kidneys with an unusually high level of clonality (clonal nephrogenesis).
Matching tumour and normal transcriptomes
Defining the nearest normal transcriptomic correlate of a cancer cell provides clues to its cell of origin and is essential for understanding precisely what transcriptomic changes are important to driving the disease. This is particularly important for childhood cancers, which have long been speculated to be of developmental origin. However, precise quantitative evidence of this hypothesis, as well as cellular resolution to the developmental cells involved is lacking. Previous efforts to define tumour transcriptomes have been limited by the need to pool mRNA across many cells, so called “bulk transcriptomics”, making it impossible to distinguish the cancer cell’s transcriptome from the normal infiltrate. The development of reliable, high-throughput single cell transcriptomic techniques now make it possible to define the tumour transcriptome with the same precision that whole genome sequencing has allowed us to define its genome.
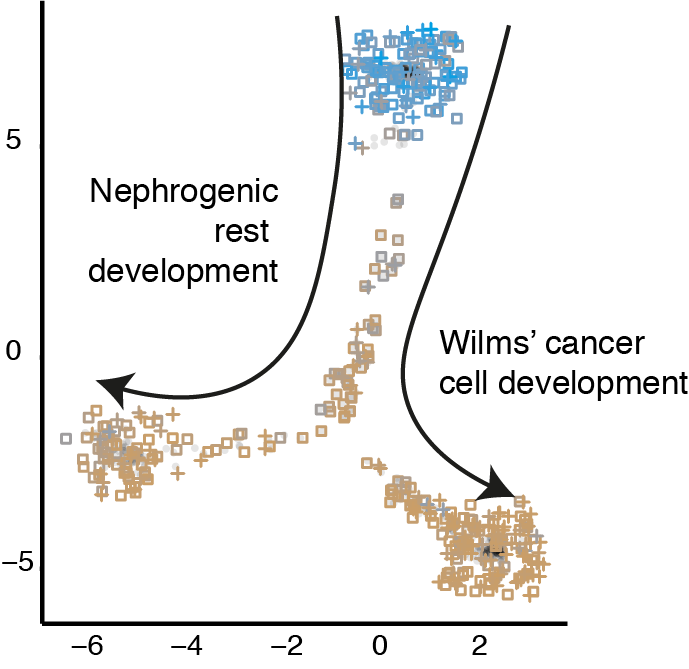 We apply these techniques to understand and define the transcriptomes of normal tissues, through our involvement in projects such as the human cell atlas. We then use the same single cell techniques to define the tumour transcriptomes of cancer cells in different tumour types. Using machine learning techniques, we compare these reference single cell atlases of normal tissue to individual cancer cell transcriptomes to identify normal cell correlates for tumour cells. For example, we used these techniques to identify the normal cell correlates of the most common types of kidney tumours and demonstrate the developmental nature of the nephroblastoma transcriptome.
We apply these techniques to understand and define the transcriptomes of normal tissues, through our involvement in projects such as the human cell atlas. We then use the same single cell techniques to define the tumour transcriptomes of cancer cells in different tumour types. Using machine learning techniques, we compare these reference single cell atlases of normal tissue to individual cancer cell transcriptomes to identify normal cell correlates for tumour cells. For example, we used these techniques to identify the normal cell correlates of the most common types of kidney tumours and demonstrate the developmental nature of the nephroblastoma transcriptome.
Clinically relevant features of transcriptomes
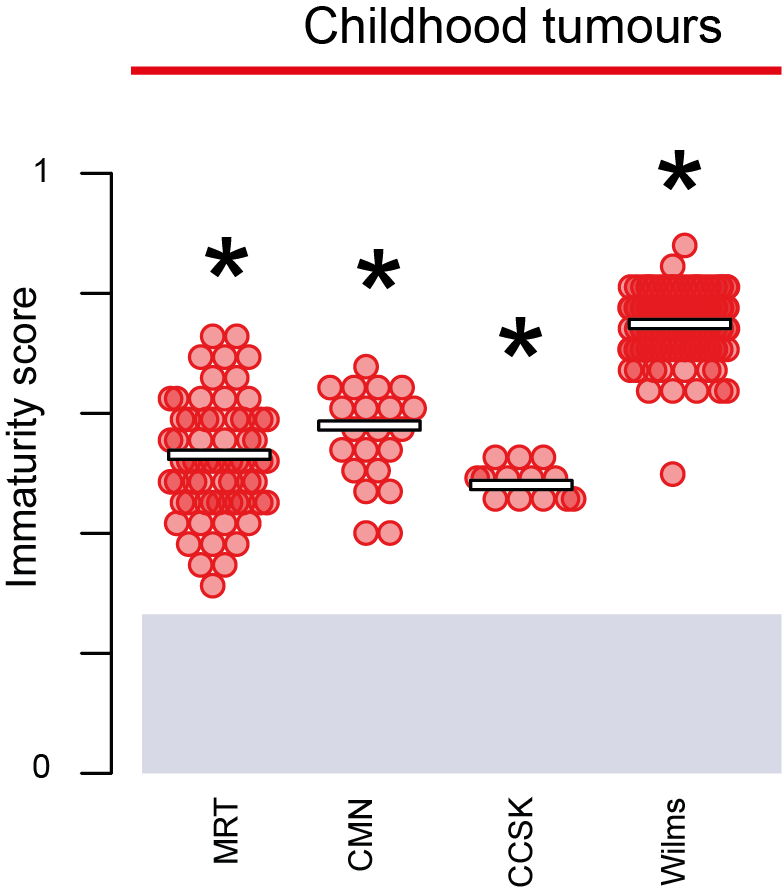
Using the above approaches, we have been able to investigate a relatively small number of samples in great detail. However, to understand the transcriptional drivers of variation with tumours and correlation clinical outcomes it is necessary to look at hundreds to thousands of tumours. Presently, this scale is not possible with single cell transcriptomic approaches, although this will change as techniques improve and costs decrease. Using statistical methods developed in the lab, we can leverage reference single cell atlases to gain insight into the “cellular signals” operative in cancer cells from bulk transcriptomes. We have used this approach to define the normal transcriptional correlate across over a thousand kidney tumours and to quantify the developmental nature of each tumour.
Join our group
We are always looking for motivated students and postdocs to join our group. Our group and the Wellcome Sanger Institute provides a friendly environment, with many opportunities to collaboration. All members of our lab work together and will be encouraged to develop their scientific, analytic, and technical skills under the supervision of Dr. Sam Behjati.
Postdoctoral fellow positions
We encourage potential postdocs to apply for independent funding. We are happy to help develop proposals and ideas.
PhD positions
To apply for a PhD position in our group, please refer to the Wellcome Sanger Institute’s PhD Programme.

Core team

Dr Nathaniel Anderson
Postdoctoral Fellow

Dr Natalie Andersson
Postdoctoral Fellow

Dr Henry Lee-Six
Group Leader (starting August 2026)

Conor Parks
Scientific Manager

Dr Lorcán Pigott-Dix
Postdoctoral Fellow

Dr Anna Wenger
Postdoctoral Fellow

Holly J. Whitfield
Postdoctoral Fellow
Previous core team members

Martin Del Castillo Velasco Herrera
Senior Bioinformatician

Dr Thomas RW Oliver
PhD Student

Tarryn Porter
Senior Scientific Manager
The following is also a member of this team:
| Sarah Farndon | Clinical Masters' student |
Associated research
Programmes and Facilities
Partners
We work with the following groups external partners