
Deciphering the Mechanisms of Developmental Disorders (DMDD)
The DMDD programme (Deciphering the Mechanisms of Developmental Disorders) is a systematic study of embryonic-lethal mouse gene lines, identifying critical genes for embryo development and survival.
About the Deciphering the Mechanisms of Developmental Disorders project
 DMDD is an international consortium of scientists who, together, have expertise in almost every aspect of embryo development. The programme’s mission is to further understanding of developmental disorders by imaging and phenotyping embryonic-lethal mouse lines. At the heart of the project is an online database, where all data is freely available to the biomedical community.
DMDD is an international consortium of scientists who, together, have expertise in almost every aspect of embryo development. The programme’s mission is to further understanding of developmental disorders by imaging and phenotyping embryonic-lethal mouse lines. At the heart of the project is an online database, where all data is freely available to the biomedical community.
Early analysis of the embryonic-lethal data has suggested links between specific genes and heart defects in the developing embryo. DMDD’s goal is that the scientific community exploits the database to the full, sparking new areas of study or broadening existing research into the genetic basis of developmental disorders.
To facilitate future research, the mouse lines are archived and are available via Infrafrontier.
Imaging
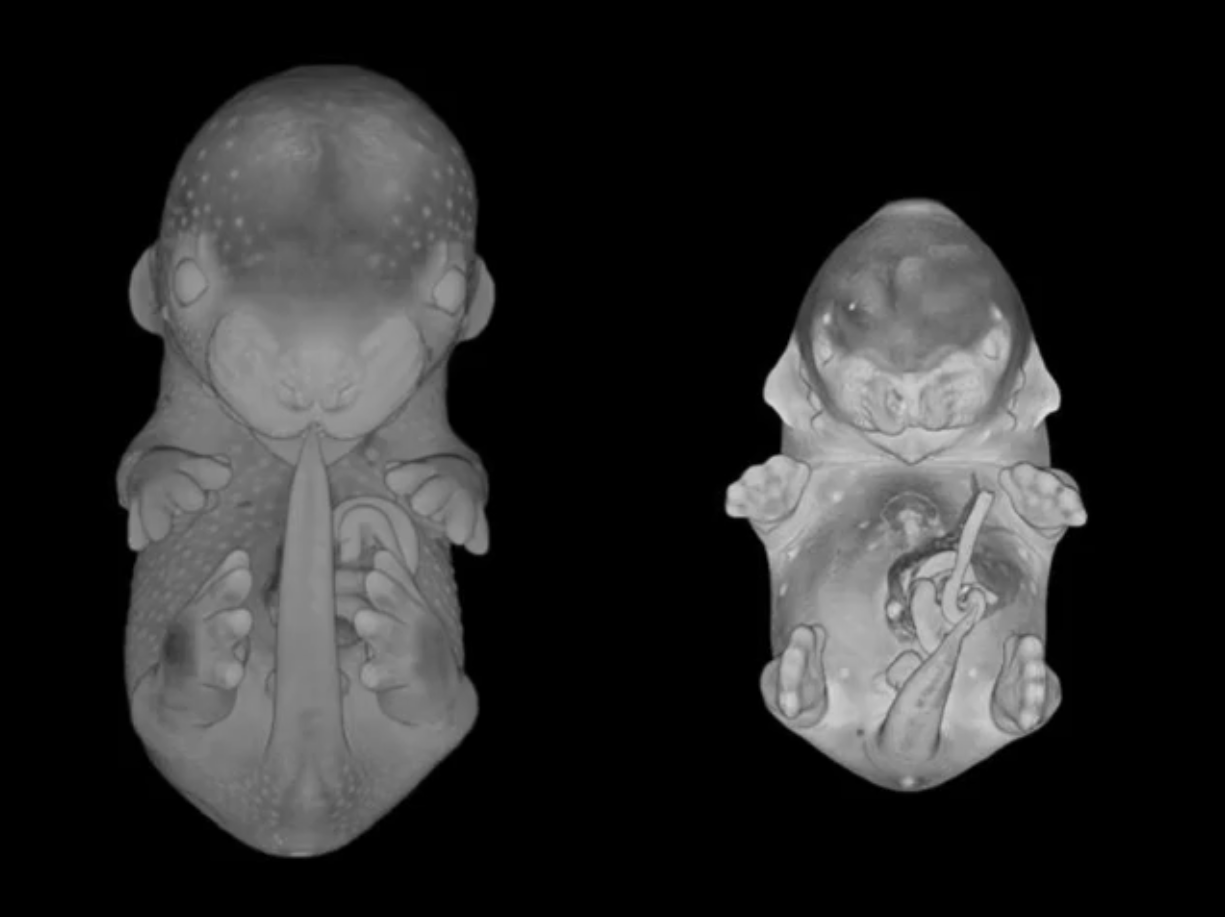
DMDD is pioneering the use of High-Resolution Episcopic Microscopy (HREM) in studies of embryonic lethal mouse lines. This technique produces images of incredibly high resolution through the whole embryo. It means that embryos with a lethal gene mutation can be compared to their wild-type siblings in exquisite 3D detail.
Phenotyping
The programme’s embryo images are studied by expert anatomists, who identify abnormalities in a range of organs and tissue structures. Placentas are also studied, in order to understand the effect of embryonic lethal gene knockouts on placental development.
Online database
All DMDD data is available to view and study online via an online database, which is an ever-expanding resource for developmental biologists and clinicians.
Embryo images can be searched by gene, phenotype or tissue, enabling researchers to identify previously unstudied genes or phenotypes relevant to their own work. A library of normal embryos is also provided for comparison.
Contact
If you need help or have any queries, please contact us using the details below.
External partners and funders
External
Wellcome Sanger Institute
At the Wellcome Sanger Institute, the Mouse Phenotyping Group produces embryos from lethal knockout gene lines and initially checks for gross morphological abnormalities. The Vertebrate Genetics and Genomics Group studies the RNA transcript of these embryos, to understand the difference in gene expression between a normal embryo and one with a gene mutation.
External
The Francis Crick Institute
The Francis Crick Institute uses high-resolution episcopic microscopy (HREM) to take high-resolution images of embryos, and is continually developing the database and web interface.
External
Medizinische Universität Wien
At the Medizinische Universität Wien, expert anatomists study the high-resolution episcopic microscopy (HREM) images and identify tissue and organ abnormalities that may have resulted from the embryonic-lethal gene knockouts.
External
Oxford University
Oxford University also phenotype embryos and are developing a method for CT scanning of embryos.
External
The UCL Institute of Child Health
The UCL Institute of Child Health studies the cardiovascular system of the embryos in detail, and provides additional phenotype information.
External
King’s College London
King’s College London considers embryos that die at or shortly after the time of birth. They examine the neural tissue in the brain and spinal cord using immunohistochemistry to understand the cause of perinatal death.
External
Babraham Institute
The Babraham Institute studies placentas from embryonic-lethal embryos using histology, in order to understand the role of the placenta in embryo development.
External
The Institute of Genetics and Molecular Medicine
The Institute of Genetics and Molecular Medicine have led the emouseatlas project, which provides an online database for normal mouse embryo anatomy and gene expression. Their detailed and annotated images of mouse embryos are a valuable reference point for comparing with embryos from embryonic-lethal lines.
External
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to improving health. We support bright minds in science, the humanities and the social sciences, as well as education, public engagement and the application of research to medicine. Our investment portfolio gives us the independence to support such transformative work as the sequencing and understanding of the human genome, research that established front-line drugs for malaria, and Wellcome Collection, our free venue for the incurably curious that explores medicine, life and art.