TRUPLEX: Enhanced Variant Identification by High-Efficiency Duplex Sequencing
Archive Page
This page is maintained as a historical record and is no longer being updated.
Technology
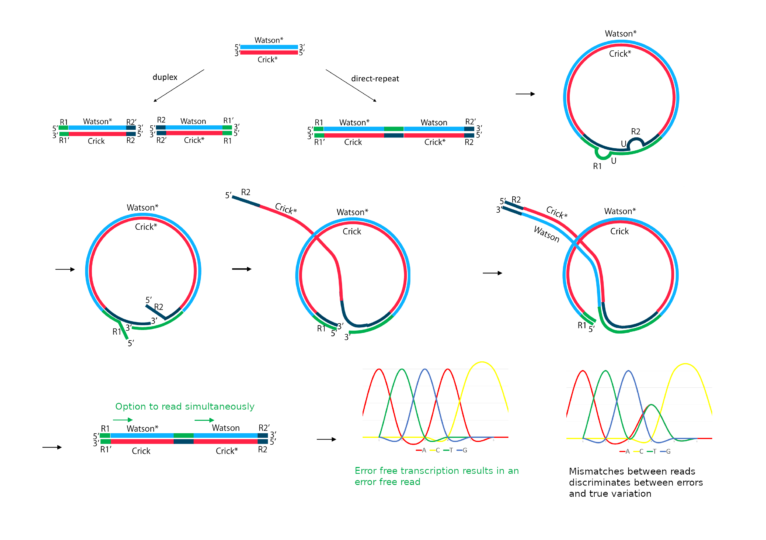
TruPlex, is a method developed by scientists at the Sanger Institute that links both DNA strands in one read so that every construct has an internal error check. This removes the need to sequence multiple PCR duplicates and dramatically reduces overall read depths required to identify variants.
TruPlex works by incorporating a first strand of DNA and the complement of its second strand into a single construct so both can be read in a sequencing by synthesis reaction. Optionally, differences between the sequence of the first and second repeats can also be read simultaneously where ambiguous bases, can be identified as low-quality base calls and discarded while the matched reads can be retained for analysis.
Advantages
- Recover data from samples where DNA availability is limited or fragmented:
– Circulating Tumour Cells/DNA
– Formalin-Fixed Paraffin-Embedded - Confidently identify rare cell population mutants in heterogeneous tissues, for example, minor cell populations in a bulk tumour.
- Efficient and easy to scale: 10-20x better yield than equivalent methods.
Detail
How it works: Direct repeats are produced by circularisation, nicking and extending the crick strand to incorporate a copy in the same molecule.
Applications/Context
Highly specific detection of rare variants. For example, monitoring minimal residual disease, identifying rare resistance mutations in cancer patients, single cell sequencing or in depth analysis of small sample biopsies.
Comparable Technologies
Current duplex sequencing methods are inefficient owing to the separate tagging and amplification of each strand. TruPlex links both strands, which are then amplified together in a single molecule. This improves efficiencies and makes the approach easier to scale.
Background
Detecting variants present at low variant allele fractions is a complicated task: library preparation and Next Generation Sequencing technologies introduce a background of errors that make it difficult to distinguish signal from noise. The ability to specifically identify variants present in one, or a few, molecules of DNA opens the possibility of detecting somatic mutations present in a small subset of cells or circulating tumour DNA.
Intellectual property
Priority patent applications filed in the US and UK.
The Wellcome Sanger Institute is offering non-exclusive diagnostic licenses to its IPR.
Download
 TruPlex_technology_flyer_JUN2020
TruPlex_technology_flyer_JUN2020
Contact
If you need help or have any queries, please contact us using the details below.
