Clean correction of a patient's genetic mutation
For the first time, scientists have cleanly corrected a human gene mutation in a patient’s stem cells. The result, reported in Nature on Wednesday 12 October, brings the possibility of patient-specific therapies closer to becoming a reality.
The team, led by researchers from the Wellcome Trust Sanger Institute and the University of Cambridge, targeted a gene mutation responsible for both cirrhotic liver disease and lung emphysema. Using cutting-edge methods, they were able to correct the sequence of a patient’s genome, remove all exogenous DNA and show that the corrected gene worked normally.
The researchers used human induced pluripotent stem cells (hiPSCs) for their research because, once reprogrammed in the Petri dish, these cells can be converted into a wide range of tissues. If stem cells from a patient with a gene defect can be corrected, scientists believe that when reintroduced into the patient they could treat the effects of the mutation causing the disease. To make such a hope reality, efficient methods of introducing DNA, repairing the gene, removing all foreign DNA and verifying the changes are needed.
For this research, the team looked at a deficiency caused by a mutation in alpha1-antitrypsin, a gene that is active in the liver where it is responsible for making a protein that protects against excessive inflammation. People with mutant alpha1-antitrypsin cannot release the protein properly from the liver, where it becomes trapped, eventually leading to liver cirrhosis and lung emphysema as a consequence. This is the commonest known inherited disorder of the liver and lung, occurring in about one in 2000 people of North European origin.
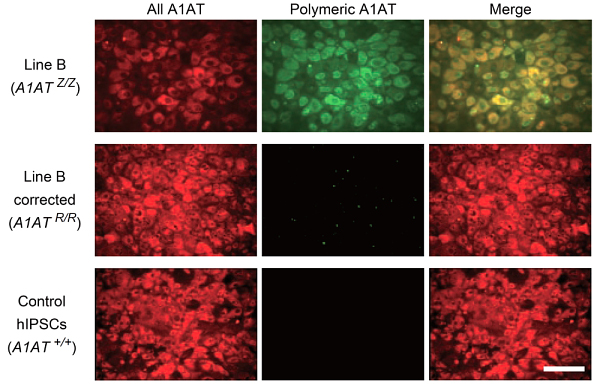
Building on previous work from Cambridge which showed that it was possible to transform skin cells into liver cells by reprogramming stem cells, the team successfully and accurately corrected an alpha1-antitrypsin gene in an established cell line containing the mutation. Using ‘molecular scissors’ to snip the genome at precisely the right place, they then inserted a correct version of the gene using a DNA transporter called piggyBac. The piggyBac sequences were subsequently removed from the cells, allowing them to be converted into liver cells without any trace of residual DNA damage at the site of correction.
The scientists then proved that the accurate copy of the gene was now active in the liver cells they had produced by demonstrating the presence of normal alpha1-antitrypsin protein in both test tube and mouse experiments.
“We have developed new systems to target genes and integrated all the components to correct, efficiently, defects in patient cells. Our systems leave behind no trace of the genetic manipulation, save for the gene correction.
“These are early steps but, if this technology can be taken into treatment, it will offer great possible benefits for patients.”
Professor Allan Bradley Director Emeritus of the Wellcome Trust Sanger Institute
“This study represents a first step toward personalised cell therapy for genetic disorders of the liver. We still have major challenges to overcome before any clinical applications but we have now the tools necessary to advance toward this essential objective.”
Dr Ludovic Vallier Medical Research Council (MRC) senior Fellow and Principal Investigator at the University of Cambridge’s MRC Centre for Stem Cell Biology and Regenerative Medicine and Department of Surgery, who studies human pluripotent stem cell biology
In their analysis of the stem cells, the research team showed that genomes of stem cells commonly contain mutations, whose cause remains unclear. However, the researchers were able to use the latest sequencing techniques to find cells with minimum numbers of mutations whose genetic consequences they could examine. They make clear that careful screening of stem cells will be needed to contribute to safe adoption of the technology.
In the final step of the project, they took cells directly from a patient with alpha1-antitrypsin deficiency and corrected the mutation exactly as they had with the established cell line. The corrected cells produced normal alpha1-antitrypsin protein.
“As there is currently no cure for this disease other than liver transplantation, and given the increasing strains being placed on the national liver transplant programme as a result of the sharp increase in the frequency of liver disease, alternative therapies for genetic and other liver diseases are urgently being sought. Our research is a critical step to developing enhancing or life-saving treatments these individuals.
“It is a quite remarkable series of results, founded on strong research and the generous participation of our patients. One of the next steps will be exploring the use of this technique in human trials.”
Professor David Lomas Professor of Respiratory Biology at the University of Cambridge and Consultant Physician at Addenbrooke’s and Papworth Hospitals (who has worked on the mechanism of alpha1-antitrypsin deficiency for 20 years and looks after patients with this condition)
More information
Funding
This work was supported by the Wellcome Trust, the Medical Research Council, the Cambridge Hospitals National Institute for Health Research Biomedical Research Center, the Papworth NHS Trust, Bill and Melinda Gates Foundation, Inserm and Institut Pasteur, as well as individual fellowships from Japan Society for the Promotion of Science, Wellcome Trust Clinical Training Fellowships and the International Human Frontiers Science Program Organization.
Participating Centres
- Wellcome Trust Sanger Institute, Hinxton, Cambridge, UK
- Laboratory for Regenerative Medicine, Department of Surgery, University of Cambridge, Cambridge, UK
- Department of Medicine, University of Cambridge, Cambridge. UK
- Innate Immunity Unit, Institut Pasteur, Inserm U668, Paris, France
- Instituto de Biomedicina y Biotecnología de Cantabria), Santander, Spain
- SangamoBioSciences Inc., Richmond, CA USA
- DipartimentoBiologia e Biotecnologie ‘Charles Darwin’, Universita’ ‘La Sapienza’, Rome, Italy
- DNAVEC Corporation, Ibaraki, Japan
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute is one of the world’s leading genome centres. Through its ability to conduct research at scale, it is able to engage in bold and long-term exploratory projects that are designed to influence and empower medical science globally. Institute research findings, generated through its own research programmes and through its leading role in international consortia, are being used to develop new diagnostics and treatments for human disease.
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to achieving extraordinary improvements in human and animal health. We support the brightest minds in biomedical research and the medical humanities. Our breadth of support includes public engagement, education and the application of research to improve health. We are independent of both political and commercial interests.


