Pig Genome
Access the map, clone and genome resources from the Porcine Genome Sequencing Project
The genome of the pig (Sus scrofa) comprises 18 autosomes, with X and Y sex chromosomes. The genome size is similar to that of human at around 2.7Gb.
About

The pig is a member of the artiodactyls, (cloven-hoofed mammal), which are an evolutionary clade distinct from the primates and rodents; there is extensive conserved homology between the pig genome and the human genome. The pig is therefore an important model for human health particularly for understanding complex traits such as obesity and cardiovascular disease. The funding for the clone based sequencing project at the Wellcome Trust Sanger Institute ran from January 2006 to December 2009.
Clone Mapping and Sequencing
A physical map of the swine genome was generated by an international collaboration of four laboratories. Both high-throughput fingerprinting and BAC end sequencing were used to provide the template for an integrated physical map of the whole pig genome.
A bacterial clone physical map of the genome was constructed using restriction enzyme fingerprinting. Fingerprints were generated by digesting clones with HindIII. Following electrophoresis on agarose gels and data collection using a fluorimager, raw images were entered using the software, IMAGE. This produced an output of normalised band values and gel traces. Analysis of data took place in WebFPC, a version of which can be found here. Clones were contiguated on the basis of shared bands. Coverage across the 2.7Gb genome was generated at 15.3x.
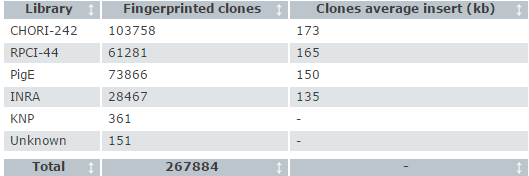
The total number of fingerprints was 267884, which were assembled into 524 contigs. These data represent the output of a stringent automated assembly, resulting in contigs of highly overlapping sets of clones, followed by initial manual editing.
Fingerprinted clones breakdown by library
 The contigs were then merged by relaxing the stringency required for overlap, as well as interrogating all other available data, such as contig localisation to the human genome via BAC end sequences, RH and genetic maps. The map, created by Sean Humphray and his team, provided a template for clone tile path selection.
The contigs were then merged by relaxing the stringency required for overlap, as well as interrogating all other available data, such as contig localisation to the human genome via BAC end sequences, RH and genetic maps. The map, created by Sean Humphray and his team, provided a template for clone tile path selection.
More information about the fingerprint map can be found in:
End sequencing
Over 600,000 BAC end sequences (BES) were generated from four libraries in three laboratories. The BES are available from the NCBI trace archive.
Using WuBLASTn set for cross-species comparison, the non-repetitive BAC ends were searched against the human reference sequence. This anchoring acted as a framework in the construction of the porcine map and along with reduced stringency fingerprint matches, enabled accelerated merging through the whole genome.
The BAC end sequencing was funded by BBSRC/DEFRA/Roslin Institute, INRA/Genoscope and the United States Department of Agriculture.
Data Availability
- NCBI Trace Archive – raw sequence data in the form of traces
- EMBL – access clone sequences via public nucleotide database
- FTP – download clone sequences via FTP
- Blast – search for clone sequences
Collaborators and Funding
Funding
The mapping and BES projects were funded by:
- USDA Cooperative State Research, Education and Extension Service (CSREES) administered the grant through the National Research Initiative
- National Pork Board
- Iowa Pork Board
- Iowa State University
- North Carolina Pork Council
- North Carolina State University
- Biotechnology and Biological Sciences Research Council (BBSRC)
- Department for Environment, Food and Rural Affairs (DEFRA)
- EU
Swine Genome Sequencing Consortium
The Sanger Institute is a member of the Swine Genome Sequencing Consortium (SGSC), a partnership of institutes involved in sequencing and genomics. The aim of the SGSC was to accelerate, facilitate and coordinate global swine genomic sequencing efforts.
Participating Centres
- Institute for Genomic Biology, University of Illinois, Urbana, IL, USA
- Department of Animal Sciences, University of Illinois, Urbana, IL, USA
- The Wellcome Trust Sanger Institute, Hinxton, UK
- Roslin Institute, Edinburgh, UK
- INRA-CEA, Jouy-en-Josas, France
- INRA-Toulouse, France
- Agricultural Research Service, Clay Center, NE, USA
- The Alliance for Animal Genomics, Bethesda, MD, USA
- University of Nevada, Reno
- Iowa State University, USA
Contact
Feedback
Whatever you want to report, comment on or ask, may it be of biological or technical nature, please use the email address below. We will either answer ourselves or forward your email to the relevant person.
If you want to report a problem, please make sure you provide as much information as possible.
Ideally we require:
- what you were looking at (URLs, database names,…)
- names, accession numbers, coordinates,…
- what exactly you were trying to do
- any error message you got
Please send your enquiries/reports/comments to: [email pig-help@sanger.ac.uk].
Downloads
Raw sequence data in the form of traces.
Access clone sequences via public nucleotide database.
Assembled sequence
Download clone sequences via FTP.
FPC takes as input a set of clones and their restriction fragments (called Bands) and assembles the clones into contigs.
Affiliated Sites
External
Roslin Institute
External
University of Illinois
External
INRA
External
NIAS Korea
External
Pig Newsletters
Data use
This sequencing centre plans on publishing the completed and annotated sequences in a peer-reviewed journal as soon as possible. Permission of the principal investigator should be obtained before publishing analyses of the sequence/open reading frames/genes on a chromosome or genome scale. See our data sharing policy.