Bacterial evolution then and there and here and now
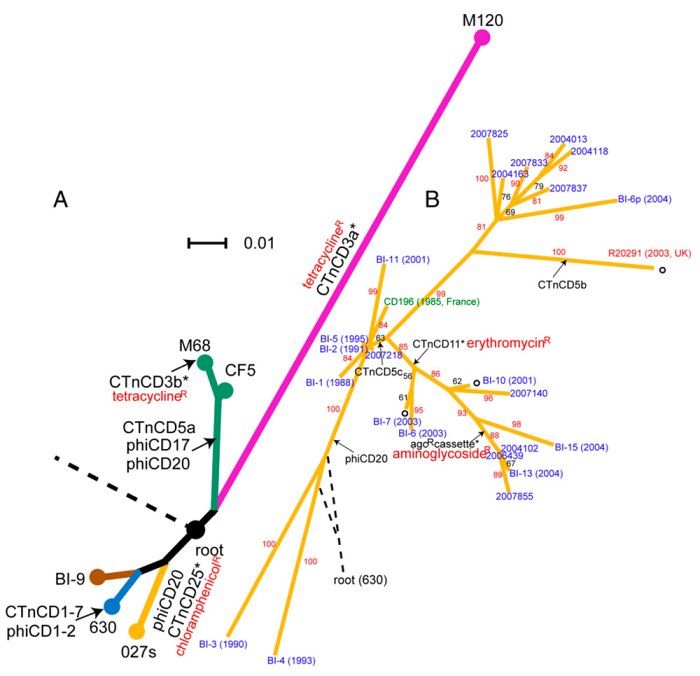
A study published today sheds new light on how Clostridium difficile (Cdiff) has emerged to cause disease in human populations. The team shows that, rather than evolving once as a disease-causing organism and spreading around the world, Cdiff has emerged as a pathogen many times and is likely to continue to do so.
The results raise the question of what is the driving force for the repeated emergence?
Cdiff becomes a healthcare threat after people have been treated with antibiotics, which disrupt the normal bugs in the gut. This allows their gut ecology to be overrun by resistant Cdiff, which produce toxins, leading to diarrhoea and gut damage. Cdiff survives and transmits easily through the bacterial family’s trademark ability to produce infectious and highly resistant spores.
It was only in 1978 that the first reports of Cdiff infections were published: in the following 30 years, it has become a global problem, with different variants emerging at different times in different countries.
To study this emergence, the team generated whole genome sequences for eight diverse Cdiff isolates, taken from six countries and including human and bovine infections. The results show that Cdiff is highly diverse; two strains of Cdiff may differ by about 2.4 per cent – greater than the divergence between chimps and humans.
Cdiff is clearly an ancient species, millions of years old, but became a significant health problem to humans only three decades ago. However, the results show that epidemic and disease-causing variants occur on several branches of this diverse tree. Its rapid emergence as a pathogen may therefore be due to changes in the environment, such as human activity, use of antibiotics or healthcare practice.
Some strains, however, have spread very rapidly. An outbreak in Canada in 2003 spread to the UK and is now responsible for 50 per cent of cases in the UK. The challenge for studying outbreaks is to identify fine detail, down to individual DNA changes, to understand how the spread has happened. New sequencing technologies make visible the previously invisible changes in many samples.
“The powerful new sequencing methods can examine macroevolution – how much do distantly collected isolates differ? – but they can also look at microevolution – how much variation occurs within an outbreak? This variation will be important in tracking the spread of these bacteria between hospitals and patients.”
Professor Julian Parkhill Senior author on the study from the Wellcome Trust Sanger Institute
To investigate the detailed picture the team sequenced the genomes of 25 isolates collected between 1985 and 2007. The data shows that genes transferred between organisms have been a powerful force in shaping the genome of Cdiff. Mobile genetic elements in bacteria often carry antibiotic-resistance genes: transfer of these elements in the Cdiff lineage has occurred repeatedly in different places at different times. Many of the genes Cdiff acquires in this way may have made it the threat it is today.
As well as the image of transfer of genes between bacteria on mobile elements, the report also looks at more familiar single-letter differences – SNPs in the genomes. Large clusters of SNPs jump out when genomes were compared with each other, highlighting regions that have undergone more than simple, single-letter changes, but result because ancestral Cdiff have exchanged regions of as much as 300,000 letters of code with more distantly related organisms from the same species, showing that Cdiff can exchange core genes across large evolutionary distances.
“We also found 12 genes under selection in the Cdiff genome: these might be candidates for those important to the infection process. Identifying genes that are selected for variation may point us to which genes are interacting directly with the host, possibly those seen by the immune system.”
Miao He First author from the Wellcome Trust Sanger Institute
Cdiff has, over recent years, risen up the healthcare agenda and the infection is now the leading cause of antibiotic-associated diarrhoeal disease – causing illness in more than 35,000 people in England’s hospitals last year. However, the genetic basis for Cdiff‘s emergence has been elusive: this work begins that archaeology.
“Disease-associated variants have emerged, and we would expect them to continue to emerge, from different lineages, in different places. This hasn’t simply happened once.
“We show how Cdiff is still evolving and expect there is more to come.”
Professor Julian Parkhill Sanger Institute
More information
Funding
This work was funded by the Wellcome Trust.
Participating Centres
- Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, United Kingdom
- Department of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, London, United Kingdom
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to achieving extraordinary improvements in human and animal health. We support the brightest minds in biomedical research and the medical humanities. Our breadth of support includes public engagement, education and the application of research to improve health. We are independent of both political and commercial interests.


