CNVs close up
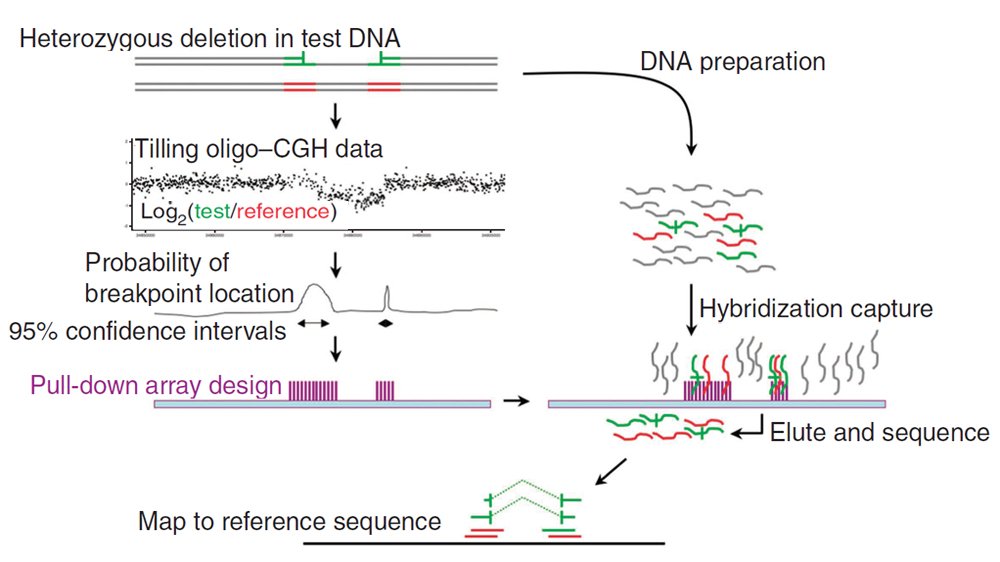
A study looking at the DNA of three people has used a cunning new approach to examine in closer detail than previously possible the mutational processes underlying copy number variation – or CNVs – in the human genome. The findings shed light on the genetic pathways behind one of the most enigmatic forms of genetic mutation in people.
Structural variation comes in many forms – missing or added chunks of DNA and sections reversed or moved around are a feature of all human genomes. Today’s publication uses a new method to take a magnifying glass to those boundaries in the genome where the sequence has been shorn or brought together in the creation of a structural variant – regions known as DNA breakpoints.
“In a single experiment, we have sequenced more CNV breakpoints than have been reported in any previous study. We developed DNA chip to isolate CNV breakpoints and then sequenced those breakpoints at absolute resolution – down to the single base pair. This should tell us more about how CNVs arise in our genomes, but it will also open the door to understand better how CNVs shape our physical characteristics and should allow us to develop ways of developing improved assays in the future.”
Dr Matt Hurles Wellcome Trust Sanger Institute researcher and lead author on the study
Although finding CNV breakpoints is crucial in developing understanding the role of structural variation in human health and disease, less than 10 per cent of known CNVs are currently characterised at singe base pair resolution.
The team looked in fine detail at more than 300 CNV breakpoints, most of which are boundaries of DNA deletions.
“Looking at the deletions, we found two predominant ‘signatures’ of mutation at the breakpoints where the DNA has been shorn. The first occurs in one third of cases, where deletions were accompanied by small stretches of inserted sequence – up to 367 letters of DNA sequence. The second signature was present in around 70 per cent of cases – here we detected that the shorn ends at either side of the deletion were identical by up to 30 bases. This signature is often referred to as ‘microhomology’.
“In the majority of cases, we identified one but not the other of these two signatures – suggesting that there are at least two distinct types of mutational processes giving rise to CNVs.”
Don Conrad Sanger Institute researcher and first author on the study
The team looked in closer detail at those cases where sequence had been inserted. Investigating 20 of the larger insertions identified, they found that in the majority of cases, the stretch of DNA that had been inserted was, in fact, a shard of DNA from the nearby genomic region.
The team also found a minority of breakpoints were simple blunt breaks.
As well as revealing a new level of detail in the mutational processes behind the development of CNVs, mapping to single base resolution allows teams to assess the functional impact of a CNV. With the right tools in place, teams can look for overlaps between CNVs and functional areas in the genome and ask: what is likely to be the impact of disruptions to the function of those particular sequences?
“This is an extremely exciting development. In the past, the technology available has been a barrier to looking at CNV breakpoints in the kind of detail we wanted to. But there is also still some way to go. Genome wide resequencing is now a reality – but it is still challenged by this type of variation (CNVs) and needs to be extended to cope with large numbers of samples. To really make inroads into understanding CNVs will require looking at far more individual human genetic material from large projects, such as the 1000 Genomes Project. This initial glimpse is incredibly interesting and certainly motivates us to want to know more!
“That will involve sampling different people; different tissues; different organisms.”
Jim Lupski Professor of Molecular and Human Genetics at Baylor College of Medicine, in Houston, Texas, USA
Ultimately, researchers hope that looking at the relative contribution of different genetic pathways to both disease causing and seemingly harmless CNVs will provide a better understanding of how structural variation can shape human evolution, health and disease.
More information
Funding
This work was supported by the Wellcome Trust.
Participating Centres
- Wellcome Trust Sanger Institute, Hinxton, Cambridge, UK
- Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts, USA
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to achieving extraordinary improvements in human and animal health. We support the brightest minds in biomedical research and the medical humanities. Our breadth of support includes public engagement, education and the application of research to improve health. We are independent of both political and commercial interests.


