Making the Most of Genetic Data
Researchers have developed new methods to mine reliably the increasing collections of genetic data for the influence of copy-number variation (CNV). The extent of CNV in the human genome, in which regions of DNA might be deleted from or duplicated in different people, has only been appreciated in the past two or three years.
The new tools can identify reliably CNVs that influence disease risk and will make collections of genetic data a much richer source of understanding.
Copy number variation (CNV) is pervasive in the human genome, and can play a causal role in genetic diseases, yet the functional impact of CNV cannot be fully captured through current methods of analysis.
“We want to integrate our new-found knowledge of structural variants into human disease studies, but off-the-shelf analysis tools can lead to false positive associations. There are no appropriate statistical methods that capture this newly appreciated variation. It is new genetic territory and we desperately needed now tools to uncover the role of CNVs.”
Dr Matt Hurles Investigator from the Wellcome Trust Sanger Institute and senior author on the study
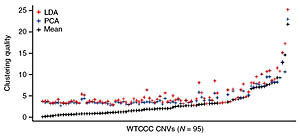
The team first examined the performance of six methods to link CNVs to disease. From these studies, they developed a new method that maximizes the power to detect real disease-causing variants, while reducing the number of false positives.
The new software system, called CNVTools, can use results from the current methods for detecting CNVs and so will be widely applicable. It can also can perform genome-wide scans involving up to 10,000 individual CNV targets.
“Often, researchers want to increase the power and specificity of their search for important variants by combining studies from different populations obtained using different methods. Our tool allows them to do that and to make allowance for the differences between CNV-detection methods.
“It will be a real benefit to large-scale studies of the type on which we and others are embarking to uncover these important but, until recently, under-appreciated types of variant. This is a new area of discovery and we need robust, high-quality methods to analyse the data.”
Dr Matt Hurles Sanger Institute
The method can reliably identify CNVs that influence disease risk as well as those that affect quantitative traits, such as height.
The CNVTools software is available from http://cnv-tools.sourceforge.net/.
More information
Funding
This research was funded by the Wellcome Trust, the Juvenile Diabetes Research Foundation and the National Institute of General Medical Sciences.
Websites
- Juvenile Diabetes Research Foundation/Wellcome Trust Diabetes and Inflammation Laboratory, Cambridge – https://www.cimr.cam.ac.uk/
- Mathematical Genetics and Statistics, Oxford – http://mathgen.stats.ox.ac.uk/
- CNVTools software – http://cnv-tools.sourceforge.net/
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to achieving extraordinary improvements in human and animal health. We support the brightest minds in biomedical research and the medical humanities. Our breadth of support includes public engagement, education and the application of research to improve health. We are independent of both political and commercial interests.


