Consortium Publishes Phase II Map of Human Genetic Variation
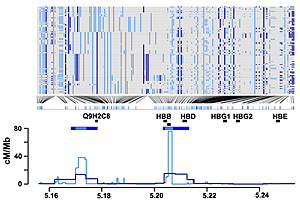
Top panel: Haplotypes from YRI in a 200 kb region around the beta-globin (HBB) gene. SNPs typed in Phase I are shown in dark blue: additional SNPs from Phase II HapMap are shown in light blue. Bottom panel: Recombination rates (lines) and the location of hotspots (horizontal blue bars) estimated for the same region from the Phase I (dark blue) and Phase II HapMap (light blue) data. Also shown are the location of genes within the region (grey bars) and the location of the experimentally verified recombination hotspot at the 5′ end of the HBB gene (black bar).
The International HapMap Consortium today published analyses of its second-generation map of human genetic variation, which contains three times more markers than the initial version unveiled in 2005. In two papers in the journal Nature, the consortium describes how the higher resolution map offers greater power to detect genetic variants involved in common diseases, explore the structure of human genetic variation and learn how environmental factors, such as infectious agents, have shaped the human genome. The first phase of HapMap is already revolutionising our ability to study the genetic basis of human disease.
Any two humans are more than 99 per cent the same at the genetic level. However, it is important to understand the small fraction of genetic material that varies among people because it can help explain individual differences in susceptibility to disease, response to drugs or reaction to environmental factors. Variation in the human genome is organized into local neighbourhoods, called haplotypes, that usually are inherited as intact blocks of information. Consequently, researchers refer to the map of human genetic variation as a haplotype map, or HapMap.
The Phase II HapMap contains more than 3.1 million genetic variants, called single nucleotide polymorphisms (SNPs) – three times more than in the initial version. The more SNPs that are on the map, the more precisely researchers can focus their hunts for genetic variants involved in disease. The rapid growth of genome-wide association studies over the past year and half has been fuelled by the HapMap consortium’s decision to make its SNP datasets immediately available in public databases, even before the first and the second versions of the map were fully completed.
Researchers around the globe have now associated more than 60 common DNA variants with risk of disease or related traits, with most of the findings coming in the past nine months. In the UK, for example, the Wellcome Trust Case Control Consortium looked at 14,000 cases and 3,000 shared controls, finding more than 20 variants associated with increased risk of a number of diseases, including coronary artery disease, Crohn’s disease, rheumatoid arthritis, type 1 diabetes and type 2 diabetes. The Consortium published their findings in Nature in June 2007.
“We are thrilled that the worldwide scientific community is taking advantage of this powerful new tool and we anticipate even more exciting findings in the future. The improved SNP coverage offered by the Phase II HapMap, along with better statistical methods, promises to further increase the accuracy and reliability of genome-wide association studies.”
Professor Gil McVean of the University of Oxford’s Department of Statistics and Wellcome Trust Centre for Human Genetics who co-led the analysis of Phase II HapMap and is one of two corresponding authors on the paper
One of the co-chairs of the analysis group, Professor Peter Donnelly, FRS, Director of the Wellcome Trust Centre for Human Genetics, said: “Understanding the differences between people’s genomes, and why those differences exist, is at the core of many questions in modern biomedical research. The HapMap project has transformed this area of research, giving new insights into areas as diverse as why some people are more susceptible to disease, and our evolutionary history.”
In its overview paper in Nature, the consortium estimates that the Phase II HapMap captured 90 to 96 per cent of common genetic variation in the populations surveyed. The consortium also confirmed that use of Phase II HapMap data has helped to improve the coverage of various commercial technologies currently being used to identify disease-related variants in genome-wide association studies.
“Thanks to this consortium’s pioneering efforts to map human genetic variation, we are already seeing a windfall of results that are shedding new light on the complex genetics of common diseases.
“This new approach of genome-wide association studies has recently uncovered new clues to the genetic factors involved in type 2 diabetes, cardiovascular disease, prostate cancer, multiple sclerosis and many other disorders. These results have opened up new avenues of research, taking us to places we had not imagined in our search for better ways to diagnose, treat and prevent disease.”
NHGRI Director Dr Francis Collins
Researchers did note However, that current technologies tend to provide better coverage in non-African populations than in African populations because of the greater degree of genetic variability in African populations. To provide information on less common variations and to enable researchers to conduct genome-wide association studies in additional populations, there are plans to extend the HapMap even further. Among the populations donating additional DNA samples are: Luhya in Webuye, Kenya; Maasai in Kinyawa, Kenya; Tuscans in Italy; Gujarati Indian in Houston; Chinese in metropolitan Denver; people of Mexican ancestry in Los Angeles; and people of African ancestry in the southwestern United States.
The overview paper also reports that the Phase II HapMap has provided new insights into the structure of human genetic variation. One new finding was the surprising extent of recent common ancestry found in all the population groups. Taking advantage of the map’s increased resolution, the researchers identified stretches of identical DNA between pairs of donor chromosomes and then compared these stretches both within and across individuals. Their analysis showed that 10 to 30 percent of the DNA segments analyzed in each population showed shared regions indicating descent from a common ancestor within 10 to 100 generations.
In addition, the new map enabled researchers to quantify more precisely the rates of shuffling, or recombination, seen among different gene classes in the human genome. In their overview paper, researchers report that recombination rates vary more than six-fold among different gene classes. The highest rates of recombination were found among genes involved in the body’s immune defence, while the lowest rates appear among genes for chaperones, which are proteins that play a crucial role in making sure other proteins are folded properly. In general, genes that code for proteins associated with the surface of cells and external functions, such as signalling, were found to be more prone to recombination than those that code for proteins internal to cells.
While the reasons for the varying recombination rates remain to be determined, the findings pose interesting evolutionary questions. In their paper, researchers suggest that one explanation may be that some recombinations in areas of the genome that affect responses to infectious agents or other environmental pressures may be selected for because they provide a survival advantage.
A related study appearing in the same issue of Nature describes how the enhanced map can help pinpoint pivotal changes in the human genome that arose in recent history. These changes, now common among various populations worldwide, became prevalent through natural selection – meaning they were somehow beneficial to human health. Although these DNA variants may still be important, their biological significance remains largely unknown.
“Human history and the genome have been dramatically shaped by environmental factors, diet and infectious disease. The gene variants identified in our study open new windows on these evolutionary forces and provide a launching point for future biological studies of human adaptation.”
Co-first author Dr Pardis Sabeti, a postdoctoral fellow at the Broad Institute of MIT and Harvard
More information
About the HapMap
The International HapMap Consortium is a public-private partnership of researchers and funding agencies from the United Kingdom, Canada, China, Japan, Nigeria and the United States. UK researchers played a major role in the HapMap project through genetic typing during the first phase, at the Wellcome Trust Sanger Institute, and much of the analysis of both phases was undertaken at the University of Oxford.
The Phase II HapMap was produced using the same DNA samples studied in the Phase I HapMap. That DNA came from blood collected from 270 volunteers from four geographically diverse populations: Yoruba in Ibadan, Nigeria; Japanese in Tokyo; Han Chinese in Beijing; and Utah residents with ancestry from northern and western Europe. No medical or personal identifying information was obtained from the donors, but the samples were labelled by population group.
Researchers can access the Phase II map data through the HapMap Data Coordination Centre (http://www.hapmap.org), the NIH-funded National Center for Biotechnology Information’s dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and the JSNP Database in Japan (http://snp.ims.u-tokyo.ac.jp/).
Participating Centres
Researchers from more than 70 centres took part in the study. Full details can be found at the Nature website.
NHGRI is one of 27 institutes and centers at NIH, an agency of the Department of Health and Human Services. NHGRI’s Division of Extramural Research supports grants for research and for training and career development. For more, visit http://www.genome.gov/
The National Institutes of Health – “The Nation’s Medical Research Agency” – is a component of the U.S. Department of Health and Human Services. It is the primary federal agency for conducting and supporting basic, clinical and translational medical research, and it investigates the causes, treatments and cures for both common and rare diseases. For more, visit http://www.nih.gov/.
The Wellcome Trust Sanger Institute – For details see below.
The Wellcome Trust Centre for Human Genetics is one of the leading human genetics research centres in the world. The Centre’s scientific objective is to explore all aspects of the genetic susceptibility of disease. The Centre houses multi-disciplinary research teams in human genetics, functional genomics, bioinformatics, statistical genetics and structural biology.
Funding
This work was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Wellcome Trust, Nuffield Trust, Wolfson Foundation, UK EPSRC, Genome Canada, Génome Québec, the Chinese Academy of Sciences, the Ministry of Science and Technology of the People’s Republic of China, the National Natural Science Foundation of China, the Hong Kong Innovation and Technology Commission, the University Grants Committee of Hong Kong, the SNP Consortium, the US National Institutes of Health (FIC, NCI, NCRR, NEI, NHGRI, NIA, NIAAA, NIAID, NIAMS, NIBIB, NIDA, NIDCD, NIDCR, NIDDK, NIEHS, NIGMS, NIMH, NINDS, NLM, OD), the W.M. Keck Foundation, and the Delores Dore Eccles Foundation.
Websites
- International HapMap Consortium – http://www.hapmap.org/
- NHGRI – http://www.genome.gov/
- National Institutes of Health – http://www.nih.gov/
- Wellcome Trust Centre for Human Genetics – http://www.well.ox.ac.uk/
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust
The Wellcome Trust is the largest charity in the UK. It funds innovative biomedical research, in the UK and internationally, spending around £500 million each year to support the brightest scientists with the best ideas. The Wellcome Trust supports public debate about biomedical research and its impact on health and wellbeing.


