Sifting through the Genome Baggage
 Evolutionary forces tend to retain important DNA sequences, whilst allowing unimportant sequences to change. Consequently, protein-coding regions – only about 1.5 per cent of the human genome – are similar in all mammalian species.
Evolutionary forces tend to retain important DNA sequences, whilst allowing unimportant sequences to change. Consequently, protein-coding regions – only about 1.5 per cent of the human genome – are similar in all mammalian species.
But there is a further 3 per cent of mammalian genome sequence that does not code for protein, yet is conserved. Are these sequences important or are they merely passengers on the evolutionary journey?
A new study from an international team co-directed by researchers at the Wellcome Trust Sanger Institute and the Broad Institute, published in Nature Genetics, shows that the vast majority of the conserved non-coding (CNC) regions are not areas that fortuitously are free of mutation, but are selectively constrained in their variation. This remarkable conclusion suggests that searches in CNC regions might lead to new discoveries of clinically important variants.
“Although we were aware of CNC regions, we could not tell whether they represented areas of the human genome that were relevant to the working of our genome, or were relics that had no present importance.
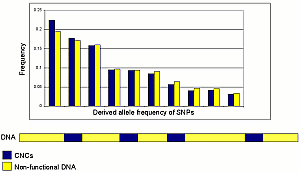
“Single-letter differences – called single nucleotide polymorphisms, or SNPs – in our genetic code are rarer in CNCs than in other, non-conserved regions. Crucially, we showed that this was not due to a lower rate of mutation, but to selection in these regions – they are under evolutionary pressure. This suggests these regions, which do not code for protein, perform important functions in our genome.”
Dr Manolis Dermitzakis Investigator, Division of Informatics at the Wellcome Trust Sanger Institute and a corresponding author
Our genome includes regulatory DNA sequences, which are important in control of genetic activity. The structure and sequence of these regions is emerging, but new methods to identify significant sequences are needed. Many of the CNC variants detected here include known regulatory regions, but also many other locations.
Finding regions of the genome where evolution has acted on variation is like finding a new pot of targets in which mutations that predispose to disease are to be discovered. The study also suggests ways in which the hunt for disease-associated variation can be made more productive.
“Our research suggests that CNCs are as important as coding sequences – but our genome has more than twice as much CNC sequence as gene sequence. This means there will be many more mutations to discover in CNCs that are associated with disease than there are in genes.
“If we include in our research a focus on these locations, we would expect to identify important variants more quickly. Our aim is to use the power of genomic information to improve our understanding of disease. This work suggests a method to harness and focus that power.”
Dr Manolis Dermatizakis Sanger Institute
Because SNPs in CNCs are relatively rare, they may not be well captured using standard methods of detecting variation (which tend to emphasize more common variants). If these regions are studied in more detail, greater biomedical benefit should follow.
More information
Corresponding Authors
- Dr Manolis Dermitzakis, Wellcome Trust Sanger Institute
- Joel N. Hirschhorn, Broad Institute of Harvard and MIT
Participating Centres
- Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, CB10 1SA, UK
- Program in Genomics and Division in Endocrinology, Children’s Hospital, Boston, MA 02115, USA
- Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, MA 02139, USA
- Department of Biomolecular Engineering, University of California Santa Cruz, CA, 95064, USA
- Division of Cardiology, Massachusetts General Hospital, Boston, MA 02114, USA
- NHLBI’s Framingham Heart Study, Framingham, MA, 01702, USA
- Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva, Switzerland
- Zoological Institute, University of Bern, Bern, Switzerland
- Department of Genetics, Harvard Medical School, Boston, MA 02115, USA
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust and Its Founder
The Wellcome Trust is the most diverse biomedical research charity in the world, spending about £450 million every year both in the UK and internationally to support and promote research that will improve the health of humans and animals. The Trust was established under the will of Sir Henry Wellcome, and is funded from a private endowment, which is managed with long-term stability and growth in mind.


