Genome of a monkey human-malaria parasite
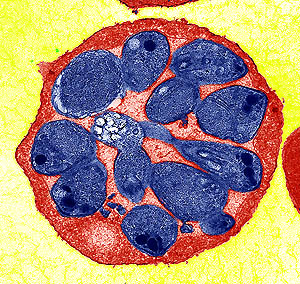
Researchers have decoded the genome of a malaria parasite that has a host range from monkeys to man. Identified originally in monkeys, the parasite was first reported in a human infection just over 40 years ago.
Until recently, four species were held responsible for human malaria infections: P. falciparum, P. vivax, P. ovale, and P. malariae. P. knowlesi is increasingly recognised as the fifth and emerging human malaria parasite, which is particularly prevalent in South East Asia and can cause potentially life-threatening malaria. Recent surveys suggest that many P. knowlesi infections have been misdiagnosed by microscopy as P. malariae, resulting in gross underestimates of its prevalence.
The genome sequence reveals a dramatic example of ‘molecular mimicry’ that is likely to be crucial for survival and propagation of the parasite in the body. Remarkably, the team found several members of a large gene family that contain sequence signatures that closely resemble a key human gene involved in regulation of the immune system. The parasite versions of the human protein are thought to interfere with recognition of infected red blood cells.
In addition to this uniquely expanded group of genes, P. knowlesi has a fundamentally different architecture of the genes involved in ‘antigenic variation’ compared to other malaria parasites. The study also emphasizes the fact that, although 80 per cent of genes are shared among all sequenced malaria parasites, each species may have a unique set of tricks and disguises that help it to escape host responses and to keep itself ahead in the host-parasite interaction.
“P. knowlesi has thrown up several surprises. Our study demonstrates the power of sequencing additional malaria genomes to unravel as yet undiscovered and fascinating aspects of the biology of malaria parasites.
“Unusually, the key genes that we think help the parasite to evade detection and destruction by host defences are scattered through the genome. In the other species we have examined, these genes are most often near the tips of the chromosomes.”
Dr Arnab Pain The first author in the study and the project manager at the Wellcome Trust Sanger Institute
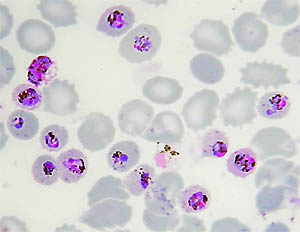
The phenomenon of ‘antigenic variation’ – where the parasite constantly changes the coat of parasitized red cells in order to avoid recognition by the host – was also first discovered in P. knowlesi. Moreover, it can be studied and grown in the lab, making it ideal to understand its basic biology such as how it invades red cells.
Identified initially as a monkey parasite, P. knowlesi had been identified in only two cases of human infection before 2004. However, at that time, Professor Balbir Singh and colleagues developed DNA-based detection methods and examined samples from malaria patients in Malaysia. They showed that almost all cases of what was thought to be infection with the human parasite P. malariae were due to infection with the ‘monkey’ parasite P. knowlesi.
“Rapid and appropriate treatment is vital in cases of malaria, but before the development of molecular detection methods, we had been hampered by our inability to distinguish between P. knowlesi and the benign P. malariae parasites by microscopy. This parasite multiplies rapidly and can cause fatal human infections, so it is vital that doctors are aware that P. knowlesi is the fifth cause of human malaria.
“The genome sequence of what has been considered to be a ‘model’ for human malaria becomes much more significant with our findings of the widespread distribution and high levels of human infections with P. knowlesi.”
Professor Balbir Singh Director of the Malaria Research Centre at the Faculty of Medicine and Health Sciences, University Malaysia Sarawak
P. knowlesi is an important model for studying the way that malaria parasites interact with host cells. It is a robust species in which invasion of red blood cells can be examined in detail. The genome sequence provides an updated catalogue of proteins that might help the parasite in these first stages of infection: the team identified novel regions in the genome that help to understand the regulation of these key genes and the transport of their proteins to the red cell surface.
Switching of surface proteins is a key defence mechanism for malaria parasites, as well as being essential for successful transfer between human and mosquito host, but the mechanisms of switching remain unclear.
“This is our first view of a monkey malaria parasite genome. It brings us intrigues and surprises – as well as new resources to help in the fight against malaria. P. knowlesi is closely related to the second-most common cause of human malaria, P. vivax. With our new understanding of the genetic architecture of both parasites, we will more efficiently translate our studies on P. knowlesi to other human parasites.
“Just as important, the genome will help in understanding human cases of knowlesi malaria.”
Dr Alan Thomas Chairman of the Department of Parasitology, Biomedical Primate Research Centre in RIJSWIJK, Netherlands

It is thought that P. knowlesi is a zoonotic malaria parasite that is transmitted by mosquitoes of the Anopheles leucosphyrus group that feed on humans and monkeys.
The function of the majority of Plasmodium proteins remains unknown. Comparison with the other malaria parasites will help to understand the differences in pathology and the mechanisms they share in interacting with the human, monkey or mosquito hosts.
The current work is published in Nature along with a companion study, deciphering the genome of another human malaria parasite Plasmodium vivax. That study was led by Dr Jane Carlton and her colleagues at the New York University School of Medicine (formerly at The Institute for Genomic Research of Rockville, Maryland, USA). The Sanger Institute is also sequencing the remaining two human-infecting Plasmodium species. The genome of P. falciparum was deciphered in 2002.
More information
Funding
This study was funded by the Wellcome Trust through its support to the Pathogen Sequencing Unit at the Wellcome Trust Sanger Institute. Part of this work was supported by the Netherlands Organization for Scientific Research, US National Institutes of Health and European Commission Framework 6 Programmes BioMalPar and the Virimal. The work was supported by the Wellcome Trust Sanger Institute Core Sequencing and Informatics Groups.
Participating Centres
- Wellcome Trust Sanger Institute, Genome Campus, Hinxton, Cambridgeshire, UK
- Ancient DNA and Evolution Group, Department of Biology, University of Copenhagen, DK-2100 Copenhagen, Denmark
- Department of Parasitology, Biomedical Primate Research Centre, 2280 GH, Rijswijk, The Netherlands
- Machine Learning Group, Department of Engineering, University of Cambridge, Cambridge, UK
- Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK
- Emory Vaccine Center, Yerkes National Primate Research Center, Emory University, Atlanta, Georgia, USA
- School of Biological Sciences, University of Liverpool, Liverpool, UK
- Institute of Biomedical and Life Sciences and Wellcome Centre for Molecular Parasitology, University of Glasgow, Glasgow, UK
- Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, Massachusetts, USA
- UBC Bioinformatics Centre and Department of Computer Science, University of British Columbia and Department of Medical Genetics, Vancouver, BC, Canada
- The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia
- The Department of Medical Biology, The University of Melbourne, Parkville, Victoria, Australia
- The Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, UK
Publications:
Selected websites
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute, which receives the majority of its funding from the Wellcome Trust, was founded in 1992. The Institute is responsible for the completion of the sequence of approximately one-third of the human genome as well as genomes of model organisms and more than 90 pathogen genomes. In October 2006, new funding was awarded by the Wellcome Trust to exploit the wealth of genome data now available to answer important questions about health and disease.
The Wellcome Trust
The Wellcome Trust is a global charitable foundation dedicated to achieving extraordinary improvements in human and animal health. We support the brightest minds in biomedical research and the medical humanities. Our breadth of support includes public engagement, education and the application of research to improve health. We are independent of both political and commercial interests.


