PoGo
PoGo – Fast Mapping of Peptides to Genomic Coordinates for Proteogenomic Analyses
Downloads
PoGo command line executables for Windows, Mac and Linux systems can be downloaded here. Additionally, a grapical user interface (GUI) for ease of use of PoGo can be downloaded as JAR file here.
To enable the successfull execution of PoGo through PoGoGUI Java 7 or higher is required and can be downloaded here.
The C++ source code for PoGo is available on github. The Java source code for PoGoGUI is available on github.
Further information
PoGo uses transcript translations and reference gene annotations to identify the genomic loci of peptides and post-translational modifications. Multiple occurrences of peptides in the input data resulting in the same genomic loci will be collapsed as a single occurrence in the output.
Input format
The input format required by PoGo is a tab delimited file with four columns. Allowed file extensions for this format are *.pogo, *.txt, and *.tsv.
| Column | Column header | Description |
|---|---|---|
| 1 | Sample | Name of sample or experiment |
| 2 | Peptide | Peptide sequence with PSI-MS nodification names in round brackets following the mpdified amino acid, e.g. PEPT(Phopsho)IDE for a phosphorylated threonine |
| 3 | PSMs | Number of peptide-spectrum matches (PSMs) for the given peptide |
| 4 | Quant | Quantitative value for the given peptide in the given sample |
Output formats
BED
This format contains the genomic loci for peptides, the exon-structure, the peptide sequence, as well as a colour code for uniqueness of peptides within the genome.
| Colour | Description |
|---|---|
| Peptide is unique to single gene AND single transcript | |
 |
Peptide is unique to single gene BUT shared between multiple transcripts |
 |
Peptide is shared between multiple genes |
PTMBED
Two files will be generates. Similar to BED the first file contains the location of the post-translational modification on the genome. Thick parts of the peptide blocks indicate the position of the post-translational modification on a single amino acid (short thick block) while longer blocks indicate the occurrence of the first and last post-translational modification and residues in between. In the PTMBED the colour code is changed to indicate the type of modification. The second file contains peptides from the input file that were not found without any post-translational modifications.
| Colour | Post-translational Modification |
|---|---|
 |
Phosphorylation (phospho) |
 |
Acetylation (acetyl) |
 |
Amidation (amidated) |
 |
Oxidation (oxidation) |
 |
Methylation (methyl) |
 |
Ubiquitinylation (glygly; gg) |
 |
Sulfation (sulfo) |
 |
Palmitoylation (palmitoyl) |
 |
Formylation (formyl) |
 |
Deamidation (deamidated) |
 |
Any other post-translational modification |
GTF
This output format contains besides the genomic loci the annotated information for the genes giving rise to each peptide sequence including status and biotype. For each mapped peptide the sample, number of peptide-spectrum matches and associated quantitative value as tags.
GCT
In this format the peptide sequences are combines with the Ensembl gene identifier. It contains the genomic loci for each peptide as well as the quantitative values for each peptide in different samples as a matrix.
Usage for command line
PoGo is a readily compiled command line tool written in C++, basic usage to generate map peptides to corresponding genomic loci with default parameters:
Windows: PoGo.exe -fasta annotation.translation.fasta -gtf annotation.gtf -in example.pogo
Linux/Unix/Mac: PoGo -fasta annotation.translation.fasta -gtf annotation.gtf -in example.pogo
Full usage:
PoGo/PoGo.exe -fasta TRANSL -gtf ANNO -in *.pogo[,*.pogo] [-format OUTF] [-merge TRUE/FALSE] [-source SRC] [-mm NUM] [-mmmode TRUE/FALSE] [-species SPECIES]
Required arguments:
-fasta TRANSL |
Filepath for file containing protein sequences in FASTA format |
-gtf ANNO |
Gene annotation with coding sequences (CDS) in GTF format |
-in *.pogo |
Path to single input file or comma separated list of paths to input files containing peptides to be mapped with associated number of peptide to spectrum matches, sample name and quantitative value (see input file format) |
Optional arguments:
-format OUTF |
Set output format GTF, GCT, BED, PTMBED or ALL. Comma separated combination possible. Default = ALL |
-merge TRUE/FALSE |
Set TRUE to merge output of multiple input files (output will be named after last input file *_merged). Default = FALSE |
-source SRC |
Set the source name for the peptides. This is only used in the GTF output. Default = PoGo |
-mm NUM |
Number of mismatches allowed in mapping (0, 1 or 2). DEFAULT = 0 |
-mmmode TRUE/FALSE |
Set TRUE to restrict number of mismatch in kmer to 1. DEFAULT = FALSE |
-species SPECIES |
Set species using common or scientific name or taxonomy ID. Default is Human (Homo sapiens, 9606). |
Table of supported species:
| Common name | Scientific name | Taxon ID |
|---|---|---|
| C.intestinalis | Ciona intestinalis | 7719 |
| Cat | Felis catus | 9685 |
| Chicken | Gallus gallus | 9031 |
| Chimpanzee | Pan troglodytes | 9598 |
| Cow | Bos taurus | 9913 |
| Dog | Canis lupus familiaris | 9615 |
| Gorilla | Gorilla gorilla gorilla | 9595 |
| Horse | Equus caballus | 9796 |
| Human | Homo sapiens | 9606 |
| Macaque | Macaca mulatta | 9544 |
| Marmoset | Callithrix jacchus | 9483 |
| Medaka | Oryzias latipes | 8090 |
| Mouse | Mus musculus | 10090 |
| Olive baboon | Papio anubis | 9555 |
| Opossum | Monodelphis domestica | 13616 |
| Orangutan | Pongo abelii | 9601 |
| Pig | Sus scrofa | 9823 |
| Platypus | Ornithorhynchus anatinus | 9258 |
| Rabbit | Oryctolagus cuniculus | 9986 |
| Rat | Rattus norvegicus | 10116 |
| Sheep | Ovis aries | 9940 |
| Tetraodon | Tetraodon nigroviridis | 99883 |
| Turkey | Meleagris gallopavo | 9103 |
| Vervet-AGM | Chlorocebus sabaeus | 60711 |
| Zebra Finch | Taeniopygia guttata | 59729 |
Step by step
- Download annotation and translated sequences for human from GENCODE, e.g. release 25. Go to www.gencodegenes.org/release/25.html and download the GTF file containing ‘Comprehensive gene annotation’ and the ‘Protein-coding transcript translation sequences’ as Fasta file. Store and unzip both files into a folder, e.g. ${POGO_DIR}/input/
- Navigate to the folder that contains the PoGo executable (cd ${POGO})
- Execute the following command to generate gtf, gct, bed and ptmbed output for of the your input file referred to as ${Peptides.txt}
Linux/Unix: ./PoGo –fasta ./input/gencode.v25.pc_translations.fa –gtf ./input/gencode.v25.annotation.gtf –in /PATH/TO/${Peptides.txt}
Windows: .\PoGo.exe –fasta .\input\gencode.v25.pc_translations.fa –gtf .\input\gencode.v25.annotation.gtf –in \PATH\TO\${Peptides.txt} - You can load the generated BED and/or GTF files into a genome browser or create an web accessible track hub for your data through TrackHub Generator. Here the example is shown for visualisation in the UCSC genome browser.
To load the data into the browser please follow these steps:- Go to https://genome.ucsc.edu and navigate to ‘My Data’ -> ‘Custom Tracks’.
- After clicking ‘Choose File’ select the file you want to upload and submit via the ‘Submit’ button.
- You will be redirected to the ‘Manage Custom Tracks’ webpage.
- Proceed from the ‘Manage Custom Tracks’ page by selecting ‘Genome Browser’ and confirm (‘go’).
- Now you can browse the peptides mapped to their genomic loci on the reference genome.
Runtime and Memory Estimations
Runtime and required memory (RAM) for PoGo execution across different settings for inclusion of mismatches and depending on number of peptides in the input file.
| Mismatch Parameter Setting | ~1,500 peptides in Input | ~250,000 peptides in Input |
|---|---|---|
-mm 0 |
< 5 min / < 4 GB | <5 min / min 10 GB |
-mm 1 |
~5 min / < 4 GB | ~5 min / min 16 GB |
-mm 2 -mmode true |
< 15 min / min 6 GB | < 20 min / min 32 GB |
-mm 2 -mmode false |
~ 1.5 h / min 64 GB | ~ 2 h / min 160 GB |
PoGo GUI
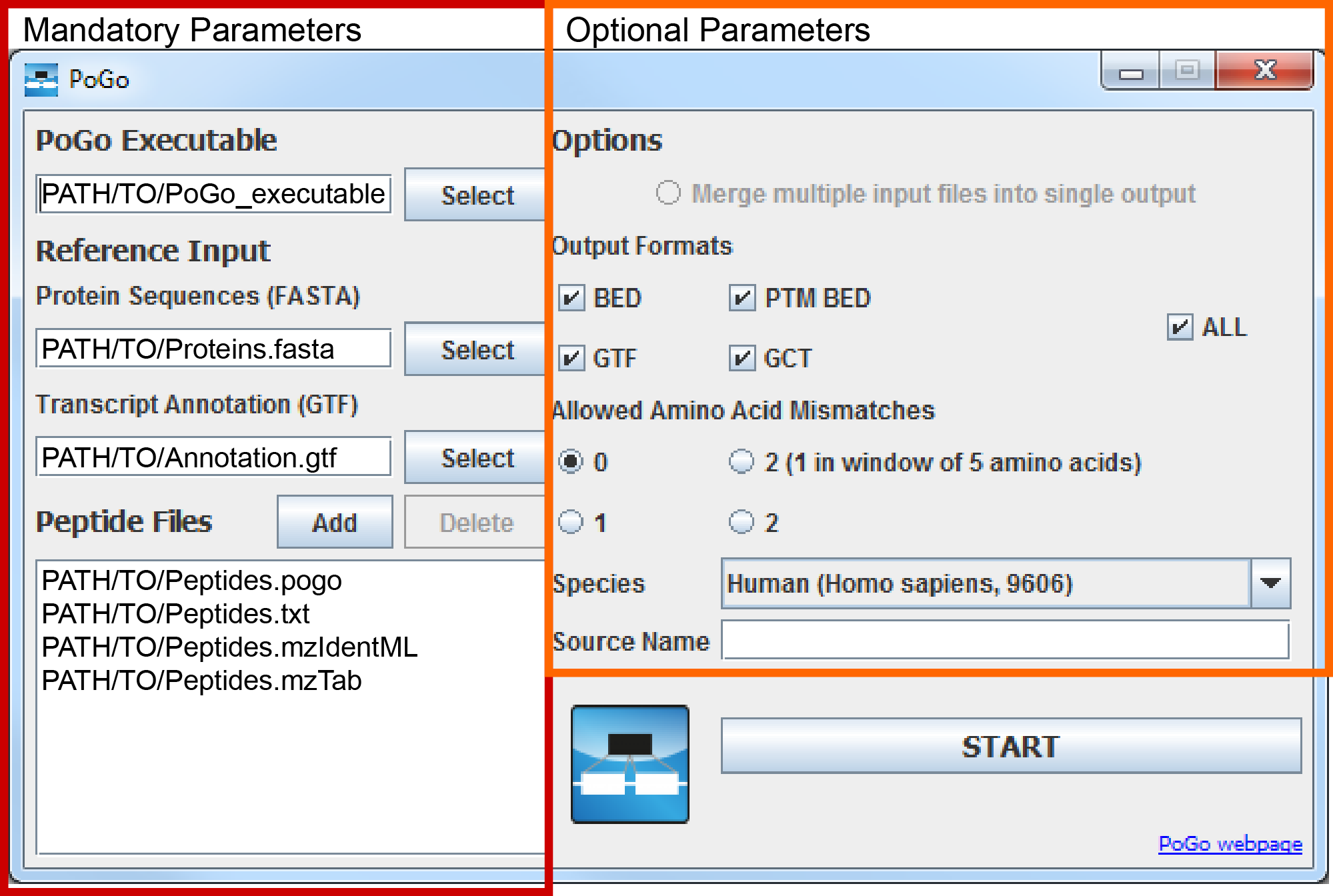
PoGoGUI uses transcript translations and reference gene annotations as well as peptide identification files in four column tab separated format, mzid, mzIdentML, and mzTab formats to pass on to the specified PoGo executable. Additional parameters to select outputformats, enable mapping with amino acid substitutions or merging output of multiple peptide identification files can be selected through checkboxes and buttons. Additional information about parameters, PoGo input and output formats can be found at the top of the ‘Learn and Support’ section.
Starting GUI
PoGoGUI has to be started through the command line. Please navitage to the folder containing the PoGoGUI.jar file via cd PATH/TO/PoGoGUI/ and type in the following command:
java -jar PoGoGUI.jar
The user interface will start. Mandatory parameters, i.e. all required files, can be chosen through the Select and Add buttons on the right half of the interface while optional parameters can be selected on the left side.
Step by Step
- Download annotation and translated sequences for human from GENCODE, e.g. release 25. Go to www.gencodegenes.org/release/25.html and download the GTF file containing ‘Comprehensive gene annotation’ and the ‘Protein-coding transcript translation sequences’ as Fasta file. Store and unzip both files into a folder, e.g. ${POGO_DIR}/input/
- Navigate to the folder that contains the PoGoGUI JAR (cd ${POGOGUI}) and execute the JAR using
java -jar ${POGOGUI}/PoGoGUI.jar - In the user interface select the PoGo executable via the selection button.
- Select the translated protein coding sequence file (FASTA) and the transcript annotation file (GTF) through the resprective Select buttons.
- Add the input files with peptides for mapping in tsv, txt, mzid, mzIdentML, mzTab formats either through the Add button or through drag and drop into the list field. The GUI will create intermediate files in PoGo input format from mzid, mzIdentML and mzTab files if required.
- Through clicking the Start button the FileConverter, if required, is started followed by PoGo.
- You can load the generated BED and/or GTF files into a genome browser or create an web accessible track hub for your data through TrackHub Generator. Here the example is shown for visualisation in the UCSC genome browser.
To load the data into the browser please follow these steps:- Go to https://genome.ucsc.edu and navigate to ‘My Data’ -> ‘Custom Tracks’.
- After clicking ‘Choose File’ select the file you want to upload and submit via the ‘Submit’ button.
- You will be redirected to the ‘Manage Custom Tracks’ webpage.
- Proceed from the ‘Manage Custom Tracks’ page by selecting ‘Genome Browser’ and confirm (‘go’).
- Now you can browse the peptides mapped to their genomic loci on the reference genome.
Test Examples
Test examples, requirement specifications and time estimations are available here.
Contact
If you need help or have any queries, please contact us using the details below.
Christoph Schlaffner (christoph.schlaffner@sanger.ac.uk)
Sanger Institute Contributors
Previous contributors

Jyoti Choudhary
Former Head of Mass Spectrometry

Christoph Schlaffner
Data Analyst