9.6 million PCR products from 50 individuals were sequenced at the Sanger Institute between 2003 and 2007 to build a more complete picture of genetic variation in human protein coding genes.
Archive Page
This page is maintained as a historical record and is no longer being updated.
Archive Page: This page is maintained as a historical archive and is no longer being updated.
About
The availability of the human genome sequence allows us a more detailed look at the variation found between individuals. The most common type of variation is the single nucleotide polymorphism (SNP), which accounts for 90 per cent of all variants. On average one nucleotide position per thousand (1 per kb) is different when two individual genomes are compared, although this figure is lower (approximately 1 per 2 kb) in protein coding sequence. Identification of all of the common coding sequence variants allows testing for biological or medical importance using genetic and functional approaches.
It is estimated that there are 11 million common variants with a minor allele frequency (m.a.f) of greater than or equal to 1 per cent, which accounts for approximately 90 per cent of the heterozygosity of the population. The remaining heterozygosity is accounted for by a very large number of other variants, each of which is very rare. When the ExoSeq project was initiated, the overall depth of sequencing coverage of the human genome was approximately five-fold (haploid n=5). This depth of sequencing has very limited power to detect the common variants of lower frequency and little chance of detecting the rare variants. The publicly available SNP resource was therefore deficient in many of the potential candidate functional variants which could be sufficiently common to make a substantial contribution to common phenotypic variation and disease.
Background
In order to generate a near-complete catalogue of common coding variants, we designed the ExoSeq project. It had the aim to re-sequence all the exons of protein coding genes: initially in 48 unrelated DNA samples from individuals of North and West European origin. The study provides a near-complete catalogue of exonic variants down to a m.a.f. of 0.03, including 95% or 62% of all variants with a m.a.f. of 0.03 or 0.01, respectively. The set includes non-synonymous changes, splice site variants, variants in UTRs, and SNPs flanking exons. Identification of rare exonic sequence variants provides valuable information about those variants that alter genome function, and those that contribute to health and disease.
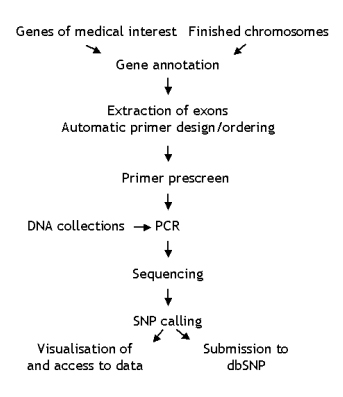
For all known protein-coding genes, novel coding sequences and transcripts, exons and their flanking sequence were extracted from theVega database, which contains high-quality manually curated annotation. Primers were designed automatically using Primer3 to amplify the exon and at least 125 base pairs either side of the exon. Any exons failing automatic primer design had primers designed manually. Primer pairs were checked for uniqueness prior to ordering and pre-screened to determine the optimum conditions for amplification. The majority of exons were amplified at 60°C. After amplification a sample of the products were visualised on an agarose gel, to confirm the size of the PCR product. The remaining PCR product was then cleaned-up using two enzymes, Exonuclease 1 and Shrimp Alkaline Phosphatase. Bi-directional sequencing of amplicons was carried out using Big DyeTM chemistry. For more details, refer to the protocols tab.
SNPs were called using ExoTrace, a novel algorithm developed in-house for the detection of heterozygotes in sequence
traces. All high-confidence variants were deposited in dbSNP on a monthly basis. An example of a candidate SNP called by ExoTrace is shown below.
We developed methods for analysing variation in other regions of the genome of potential functional importance, including promoters and other regulatory elements identified as those showing varied levels of conservation between species. By extending the study to the identification of non-coding variants we built a more complete picture of the functional consequences of variation in the human genome.
Collaborative studies of variations in disease in several areas including epilepsy, cardiovascular disease, platelet biology, deafness and genes involved in cognition were undertaken. Exons were sequenced in the standard DNA panel (as described above) and patient samples as appropriate, to identify additional SNPs for association studies or direct detection of mutations.
Protocols
Details of all ExoSeq protocols can be downloaded below:
SNP Identification
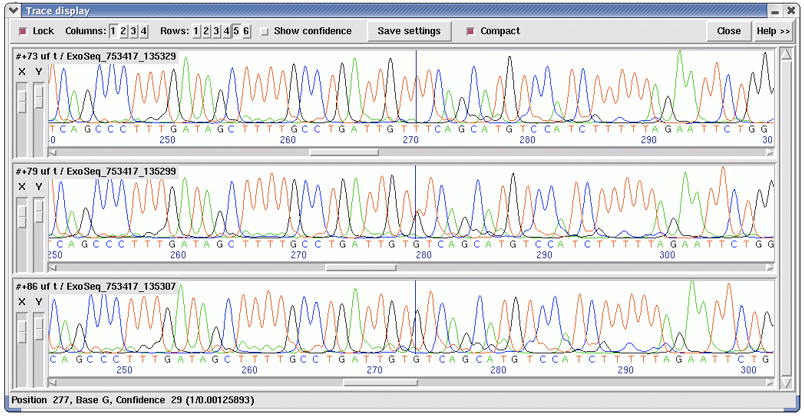
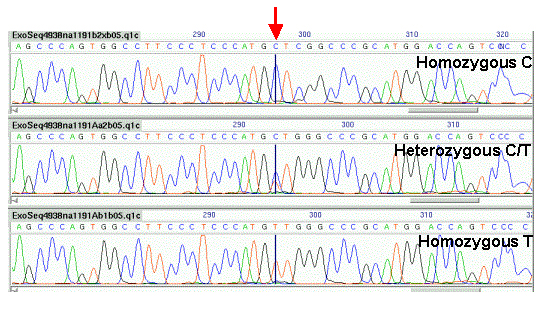
A novel SNP caller, ExoTrace, was developed for the ExoSeq project for the detection of heterozygous SNPs in sequence traces. The program worked by comparing actual peak heights with the expected peak height for a homozygous base. A base was called as homozygous if the relative peak height in a single channel exceeded a threshold and the signal in all other channels was significantly smaller than the expected peak height. A base was called as heterozygous if the signal in two channels was approximately half the expected homozygous peak height and there was no significant signal in the other channels (Figure 1). The results of the SNP calling were displayed in a specific implementation of GAP4.
Figure 1 shows an example of three sequence reads from three different individuals showing three different genotypes for a G/T model SNP indicated by the vertical black line in the middle of each trace. The top trace shows a homozygous base (T), the middle trace shows a heterozygous base (GT) and the bottom trace shows a homozygous base (G).
ExoSeq SNP Identification PDF – Please download this PDF for details about ExoTrace and SNP identification
Statistics
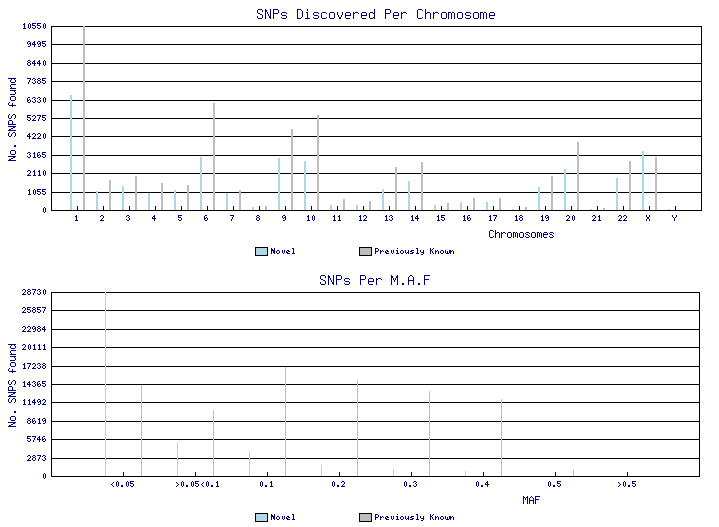
ExoSeq project results: last updated Monday 07 January 2008
- Number of SNPs identified : 89914
- Number of Novel SNPs : 34932
- Number of Non-Synonymous SNPs : 8710
- Number submitted to dbSNP : 84678
DNA Panel Samples
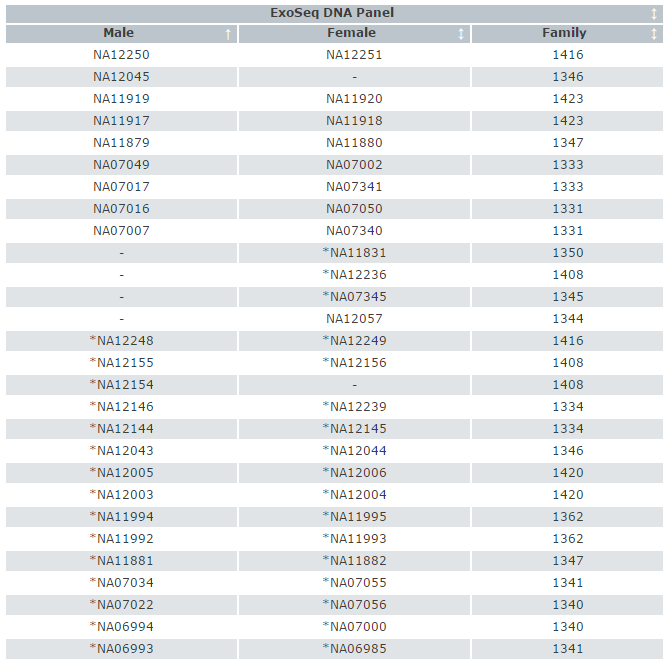
The DNA panel used for the exon sequencing project consists of 50 individuals from the CEPH families, supplied by Coriell Cell Repositories. The Coriell Cell Repository number, gender and family number for each sample is provided in the table below.
DNA panel sample preparation – Please download this PDF for details of the sample preparation.
Thirty samples are shared with HapMap (plate 1), indicated by *.
Downloads
SNPs were submitted to dbSNP on a regular basis from the results of sequencing the CEPH DNA samples as part of the core project.
Affiliated Sites
External
Vega
Data use
This sequencing centre plans on publishing the completed and annotated sequences in a peer-reviewed journal as soon as possible. Permission of the principal investigator should be obtained before publishing analyses of the sequence/open reading frames/genes on a chromosome or genome scale. See our data sharing policy.