Genome secrets of elusive human malaria species revealed
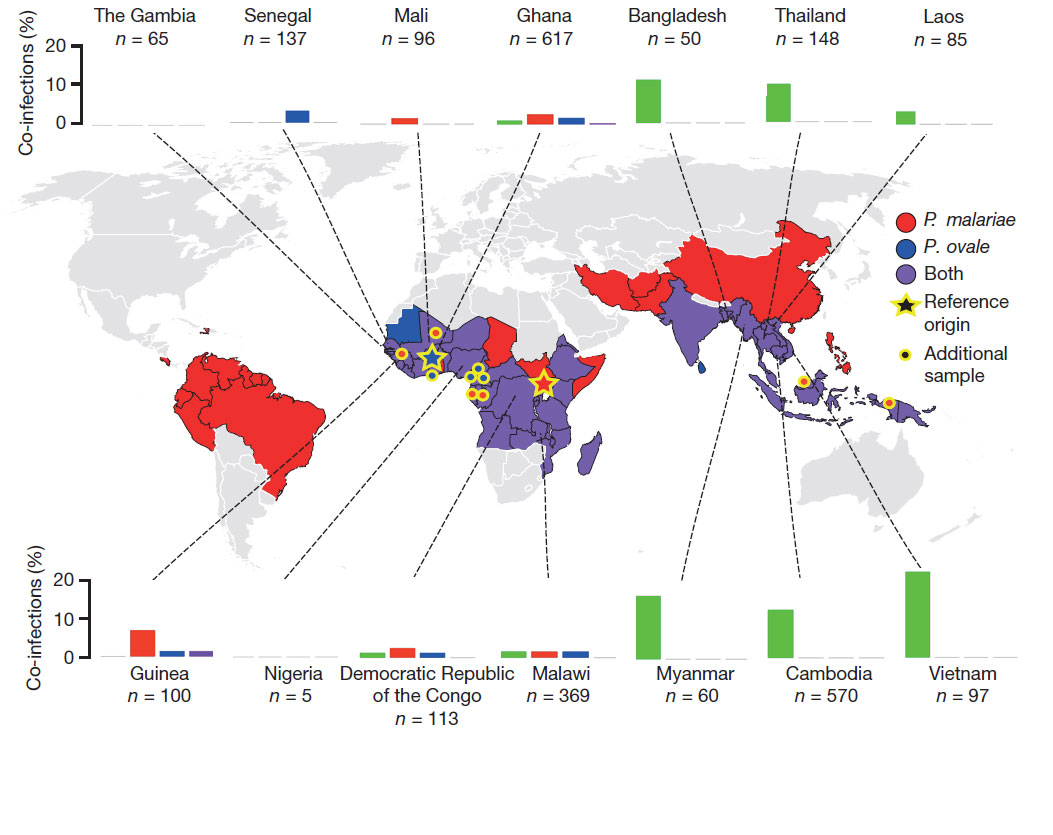
The genomes of the two least common species of human malaria parasites are revealed today (25 January 2017) in Nature by a team of scientists from the Wellcome Trust Sanger Institute and their international collaborators. These sequences will enable improved surveillance and diagnosis of these rarer parasites that still cause more than 10 million malaria cases every year.
The research has important implications for malaria eradication worldwide, and casts light on a malaria vaccine target.
Malaria is caused by Plasmodium parasites, which are spread to humans by mosquitos. The genomes of three human infective Plasmodium species are relatively well studied, especially P. falciparum, the most common malaria parasite. However, very little was known about Plasmodium malariae and Plasmodium ovale, which are believed to cause up to 5 per cent of malaria worldwide, corresponding to approximately 10 million cases annually. These species can remain hidden in the host for years.
The researchers determined the genome sequences of these Plasmodium parasite species. By comparing these new genomes with those of the malaria parasites already sequenced, the researchers were able to identify genes that could be involved in human infection and in adapting to the human host. They found that up to 40 per cent of the P. malariae and P. ovale genomes contain genes that are probably involved in evading an immune response.
The study revealed that P. malariae contains two new families of genes that are similar in shape to a vital gene in P. falciparum, known as RH5. This gene is essential for the P. falciparum parasite to invade human red blood cells and is one of the top targets for malaria vaccine design. It is likely that the novel P. malariae genes are also involved in binding to host cell receptors.
“It is really hard to study these parasites because we can’t grow them in the lab. Here, we isolated the parasites from blood samples of malaria patients and determined these final Plasmodium genome sequences. This will help us understand the evolution of the Plasmodium species, and maybe even give us an idea which routes to drug resistance these parasites may possess.”
Gavin Rutledge First author on the paper from the Wellcome Trust Sanger Institute
“Although they are less lethal than Plasmodium falciparum, the rarer malaria species are likely to be much more difficult to eliminate. Better tools to diagnose these parasites, as well as drugs and vaccines to control them will be essential. These new genomes should now make it possible to develop improved diagnostic tools for these Plasmodium species, to ensure that drugs work against them and to assist vaccine development.”
Professor James McCarthy from QIMR Berghofer Medical Research Institute
P. ovale actually consists of two distinct species Plasmodium ovale wallikeri and Plasmodium ovale curtisi. The authors showed that the split between these species was ancient and occurred long before the much more virulent P. falciparum emerged. The researchers also sequenced Plasmodium parasites taken from chimpanzees living in a sanctuary in Gabon. They compared these with the human samples, and existing data from other Plasmodium parasites that also infected chimpanzees, offering insights into how malaria parasites have adapted to different host species.
The new genetic information is already available for other scientists in the malaria research community to use via the Sanger Institute GeneDB database or the European Nucleotide Archive at the European Bioinformatics Institute.
“This study provides long awaited reference genomes for the malaria research community. The parasites are present in malaria zones worldwide yet researchers have limited knowledge about their biology. The genomes of these more neglected species will enable the development of tools to study malaria transmission and spread, which will be essential to achieve the goal of complete malaria eradication.”
Dr Thomas Dan Otto, lead author from the Sanger Institute
More information
Funding
This work was supported by the Medical Research Council, the Wellcome Trust and other funding bodies. Please see the paper for full funding information.
Participating Institutes
- Wellcome Trust Sanger Institute, Hinxton, Cambridge, United Kingdom
- Malaria Research and Training Center, University of Science, Techniques, and Technologies of Bamako, Bamako, Mali
- Bernhard-Nocht Institute for Tropical Medicine, Hamburg, Germany
- University of Buea, Buea, Cameroon
- Navrongo Health Research Centre, Navrongo, Ghana
- Centers for Disease Control and Prevention, Atlanta, Georgia, United States
- Laboratoire MIVEGEC (UM1-CNRS-IRD), Montpellier, France
- Centre International de Recherches médicales de Franceville, Gabon
- Global and Tropical Health Division, Menzies School of Health Research and Charles Darwin University, Darwin, NT, Australia
- Centre for Tropical Medicine and Global Health, Nuffield Department of Clinical Medicine, University of Oxford, UK
- Clinical Tropical Medicine Laboratory, QIMR Berghofer Medical Research Institute, Brisbane, Australia
- Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom
- Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
Publications:
Selected websites
QIMR Berghofer Medical Research Institute
QIMR Berghofer is a world-leading medical research institute in Brisbane, Australia. It has established an international reputation for research excellence and consistently rates in Australia’s top two medical research institutes. QIMR Berghofer’s research is focused on cancer, infectious diseases, mental health and chronic disorders. The institute is conducting clinical trials of promising malaria drugs using funding from Medicines for Malaria Venture (MMV).
The Wellcome Trust Sanger Institute
The Wellcome Trust Sanger Institute is one of the world’s leading genome centres. Through its ability to conduct research at scale, it is able to engage in bold and long-term exploratory projects that are designed to influence and empower medical science globally. Institute research findings, generated through its own research programmes and through its leading role in international consortia, are being used to develop new diagnostics and treatments for human disease.
Wellcome
Wellcome exists to improve health for everyone by helping great ideas to thrive. We’re a global charitable foundation, both politically and financially independent. We support scientists and researchers, take on big problems, fuel imaginations and spark debate.


